

Abstract
The mRNA of human NF-κB repressing factor (NRF) contains a long 5′-untranslated region (UTR) that directs ribosomes to the downstream start codon by a cap-independent mechanism. Comparison of the nucleotide (nt) sequences of human and mouse NRF mRNAs reveals a high degree of identity throughout a fragment of 150 nt proximal to the start codon. Here, we show that this region constitutes a minimal internal ribosome entry segment (IRES) module. Enzymatic RNA structure analysis reveals a secondary structure model of the NRF IRES module. Point mutation analysis of the module determines a short, 14-nt RNA element (nt 640–653) as a mediator of IRES function. Purification of IRES binding cellular proteins and subsequent ESI/MS/MS sequence analysis led to identification of the RNA-binding protein, JKTBP1. EMSA experiments show that JKTBP1 binds upstream to the 14-nt RNA element in the NRF IRES module (nt 579–639). Over-expression of JKTBP1 significantly enhances activity of the NRF IRES module in dicistronic constructs. Moreover, siRNA experiments demonstrate that down-regulation of endogenous JKTBP1 decreases NRF IRES activity and the level of endogenous NRF protein. The data of this study show that JKTBP1 and the 14-nt element act independently to mediate NRF IRES activity.
Keywords: IRES, JKTBP1, NRF, RNA structure, translation
INTRODUCTION
The NF-κB repressing factor (NRF) is a regulatory transcription factor that represses the basal transcription of various cytokine genes, interferon-β (IFN-β), interleukin-8 (IL-8), and inducible nitric oxide synthase (iNOS) (Nourbakhsh and Hauser 1999; Nourbakhsh et al. 2001; Feng et al. 2002). The NRF gene is located on the X chromosome in mouse and human. Two NRF encoding mRNAs of 3.7 and 2.8 kb were identified in various human cell lines (Nourbakhsh and Hauser 1999). Both mRNAs share a common 5′-untranslated region (UTR) of 653 nucleotides (nt) but differ in their 3′-UTRs. Mammalian mRNAs generally contain a shorter 5′UTR (<150 nt) and are translated by a mechanism known as cap-dependent translation initiation. This mechanism is characterized by ribosomes scanning from the capped mRNA 5′ end along the mRNA in 3′ direction until they recognize an AUG codon in a favorable context. The 5′-UTR of NRF mRNA has two features that are incompatible with efficient cap-dependent ribosomal scanning: it is predicted to fold into a complex secondary structure (−126.3 kcal/mol) with several stable hairpins, and it contains 11 short open reading frames. Indeed, we have shown that the human NRF 5′-UTR acts as an internal ribosome entry segment (IRES) in vivo (Oumard et al. 2000). Further analysis revealed that NRF IRES is inactive in vitro but acts in a distance-independent manner with respect to the AUG start codon in various cell lines (Oumard et al. 2000).
Translation initiation by internal ribosome entry was first shown for mRNAs of picornaviruses (e.g., poliovirus) (Mountford et al. 1994; Byrd et al. 2005). Later, a number of IRES modules were identified in cellular mRNAs (Vagner et al. 1995; Yang and Sarnow 1997; Stein et al. 1998; Hellen and Sarnow 2001). Similar to viral IRES modules, cellular IRES modules enable translation of the mRNAs under conditions when cap-dependent protein synthesis is impaired. Although a common functional mechanism of cellular IRES modules is not known, it is generally assumed that they require the binding of auxiliary cellular factors, called IRES trans-acting factors (ITAFs), to initiate translation at a downstream AUG codon (Komar and Hatzoglou 2005).
In order to identify proteins that regulate cap-independent translation in NRF mRNA, we first isolated a functional minimal IRES module by dissecting human and murine NRF 5′-UTRs. Further characterization of NRF IRES module identified a short 14-nt RNA element (nt 640–653) as mediator of IRES function. Using RNA electromobility shift assays (EMSA) as well as purification of NRF IRES binding proteins with following electrospray ionization and tandem mass spectrometry (ESI/MS/MS), we identified JKTBP1 as an NRF IRES binding protein. JKTBP proteins are related to a family of heterogeneous nuclear ribonucleoproteins (hnRNPs). These proteins are composed of two RNA binding domains closely related to hnRNP D/AUF1 protein (Tsuchiya et al. 1998; Kamei et al. 1999; Dean et al. 2002). Three isoforms of JKTBP (1, 2, and Δ6) are known, which are abundant in many cell lines. JKTBP1 was shown to bind to AU-rich RNA elements in 3′-UTRs of TNF-α and COX-2, suggesting a possible role in mRNA biogenesis (Dean et al. 2002). Despite its obvious affinity to RNA, the precise function of JKTBP1 was not known. Our data demonstrate for the first time that JKTBP1 contributes to cap-independent translation initiation by NRF IRES. Although acting independently, JKTBP1 and the 14-nt RNA are required to mediate NRF IRES activity.
RESULTS
Mapping of the NRF IRES module
The human NRF 5′-UTR acts as a highly efficient IRES in dicistronic constructs (Oumard et al. 2000). Human and murine cell lines show matching expression patterns and regulatory function of NRF, suggesting identical regulation (Nourbakhsh and Hauser 1999; Froese et al. 2006). More precisely, their 5′-UTRs may contain conserved functional elements that mediate internal ribosome entry in both species. We compared the nucleotide sequences of human (accession numbers EF216688 and EF216689) and murine (accession number EF165096) NRF 5′-UTRs in order to identify conserved functional elements. Four homologous regions were identified upstream of the AUG start codon (Fig. 1). To test whether these regions impart IRES function, serial truncations of both 5′-UTRs were inserted into the intercistronic region of a dicistronic luciferase construct (Fig. 1). The first cistron, encoding Renilla luciferase, is translated by a cap-dependent scanning mechanism. The second cistron, encoding firefly luciferase, requires translation by internal ribosome entry. All constructs were transfected together with CAT expression plasmid into murine C243 cells and tested for CAT, Renilla luciferase, and firefly luciferase activities. We calculated the ratio of firefly luciferase activity to Renilla luciferase activity in order to estimate a relative factor for IRES activity. IRES-dependent expression of the second cistron varies widely, whereas expression of the first cistron diverges slightly in all reporter plasmids (Fig. 2A,B). As expected, both full-length NRF 5′-UTRs show significant IRES activity. The integrity of these dicistronic mRNAs was confirmed by Northern blot analysis (Oumard et al. 2000). Successive 5′-truncation of the NRF 5′UTR of the human (nt 1–501) or mouse (nt 1–857) has no, or marginal, effect on IRES activity, but 3′ truncation of only 50 nt essentially eliminates IRES activity in pRL001–600FL and pRLmu 001–807. Strikingly, fragments bearing homologous 150-nt sections of the human (nt 501–653) or mouse (nt 708–857) NRF 5′-UTR retain significant IRES activity. Four dicistronic constructs lacking either SV40 promoter (DRLmu001–807FL and DRL001–653FL) or SV40 promoter and the Renilla luciferase (Dmu001–807FL and D001–653FL) coding region served as additional controls (Fig. 2C). All these constructs reveal a negligible level of Renilla and firefly luciferase activities. This excludes a cryptic promoter activity of human and murine NRF 5′UTRs. Together, our data strongly suggest that the 150-nt fragments (nt 501–653 of human or nt 708–857 of murine NRF 5′UTR) immediately upstream of the NRF start codon constitute minimal IRES modules.
FIGURE 1.

Comparison of human and murine NRF 5′UTR. Serial truncations of both 5′UTRs were inserted into the intercistronic region of a dicistronic luciferase construct pRLFL at the top. Lengths of linker sequences are indicated directly above the schematic diagram. Sequence comparison of human (653 nt) and murine (857 nt) NRF 5′UTRs is shown (accession numbers EF21688 and EF165096, respectively). The murine NRF sequence is shown in bold. Dots are added to obtain maximal homology. Homologous regions are shown in hatched regions together with the percent homology. Nucleotide positions are depicted with respect to the identified 5′ end cDNA of human or murine NRF (+1).
FIGURE 2.

Dissection of human and murine NRF 5′-UTRs. C243 cells were transfected with 1 μg of dicistronic constructs containing various regions of human or murine NRF 5′-UTRs and 1 μg of monocistronic CAT control plasmid (pMPSV-HECAT). Details of the transfected constructs are schematically presented at the top of Figure 1. Lengths of linker sequences are indicated directly above the schematic diagram. Dicistronic plasmids containing murine NRF 5′-UTR are designated by the abbreviation “mu.” Numbers indicate boundaries of inserted 5′-UTR respective to the 5′ end of NRF cDNAs (+1). Luciferase activity and CAT activity were determined 2 d after transfection. Luciferase activities were normalized to CAT activity. (A) Renilla luciferase activities normalized to CAT activity are shown as mean relative light units±SEM of at least five independent transfection experiments. Relative Renilla luciferase activities of cells transfected with the dicistronic plasmids containing human NRF 5′-UTR (white bars) or murine 5′-UTR (hatched bars) are shown. The relative Renilla luciferase activity of cells transfected with the dicistronic plasmid pRL001–653 containing full-length human NRF 5′-UTR in a single transfection experiment was set to 1. (B) Relative IRES activities of cells transfected with the dicistronic plasmids containing human NRF 5′-UTR (black bars) or murine 5′-UTR (shaded bars) are shown, calculated as the ratio of firefly and Renilla luciferase activities for each transfected construct in each transfection experiment. (C) DRLmu001–807FL and DRL001–653FL constructs lack the SV40 promoter, and Dmu001–807FL and D001–653FL lack both the SV40 promoter and Renilla luciferase coding region. Details of these constructs are schematically presented at the top. Details of dicistronic constructs, pRL001–653FL and pmuRL001–857FL, are schematically presented at the top of C. Renilla and firefly luciferase light units of 0.5 × 106 cells normalized to CAT activity are shown as mean value±SEM of at least three independent transfection experiments. Relative Renilla luciferase light units of cells transfected with the dicistronic and monocistronic plasmids containing human NRF 5′-UTR (white bars) or murine 5′-UTR (hatched bars) are shown on the primary axis. Relative firefly luciferase light units of cells transfected with the dicistronic and monocistronic plasmids containing human NRF 5′-UTR (black bars) or murine 5′-UTR (gray bars) are shown on the secondary axis.
Structural analysis of the NRF IRES module
It is commonly assumed that IRES modules mediate their function through specific secondary- and tertiary-structure elements. Secondary-structure prediction of the human NRF IRES module suggested the presence of two independent stem–loops at nt 640 and 653 near the AUG codon (data not shown). To derive experimentally a secondary-structure model for the human NRF IRES module, the accessibility of bases to several RNases was determined (Fig. 3A). Briefly, 5′-end-labeled NRF IRES module RNA (NI0) and a mutant variant containing modifications at nt 649–652 (NIPST0) were synthesized in vitro and incubated with RNase V1, A, or T1 (Fig. 3A, V1, A, and T1). Labeled transcripts contain 34 unrelated nucleotides corresponding to the multicloning site (MCS), nt 507–653 of human NRF 5′UTR close to NRF cDNA sequences coding for aa 1–17 (Fig. 4A). Prior to RNase treatment, both labeled NI0 and NIPST0 probes revealed a full-length RNA fragment (Fig. 3A, lanes 1,2). The identity of each nucleotide was determined by comparison with the corresponding RNA sequencing reaction using RNase T1 under denaturing conditions (Fig. 3A,S, lanes 3,4) or a ladder of alkaline hydrolyzed NI0 RNA fragments (Fig. 3A,H, lane 11). RNase V1 preferentially cleaves 5′ to a paired nucleotide. RNase A and T1 cleave 3′ to single-stranded C or U and G, respectively. It is clear that the digestion activity of RNases V1, A, and T1 on native RNA is highly sensitive to the structural context and accessibility of each nucleotide. Nevertheless, numerous nucleotides were susceptible to cleavage (Fig. 3A). Cleaved nucleotides between G577 and G657 were used to constrain the RNA folding algorithm MFOLD and generate putative secondary structures (Fig. 3A, lanes 5–10) (Zuker 2003). The weakly modified positions were detected by longer exposition times (data not shown) and used to further refine these structures. Cleavage sites downstream from nt 657 are difficult to resolve on the gel. The NRF coding region contains an unpaired stretch of 6 A nt close downstream from G657, which is not susceptible to cleavage and thereby appears as an open frame on the gel. We note that nucleotides between G617 and G614 and nucleotide U624 were weakly cleaved by RNase V1 but, more strongly, by RNase T1 and A. Thus, these nucleotides were considered to be unpaired in Mfold analysis. To simplify, the data are schematically summarized in Figure 3A (bottom). Most importantly, the region from nt 640 to nt 653 of the NRF IRES module appears to adopt a small stem–loop (I) that is indeed altered in NIPST0. In NI0, RNase V1 cleaves between nt 649 and 650, 650 and 651, 642 and 643, and 643 and 644. RNase A cleaves 3′ to nt 646 and 648 but not 3′ to nt 645. In both NI0 and NIPST0, nt 557–640 form a large stem–loop (II) strongly sensitive to RNase A and T1 and an additional small stem–loop (III) upstream. Short-run gel electrophoresis of the protected fragments revealed an indistinguishable secondary structure of NI0 and NIPST0 in the region further upstream between nt 507 and 563 (data not shown).
FIGURE 3.

Comparison of wild-type (WT) and mutant human NRF IRES modules. (A) Enzymatic structure analysis of NRF IRES module is presented. Schematic diagram of the constructs used to generate WT (NI0) or mutant (NIPST0) of NRF IRES module is shown at the top of A. Constructs were digested with NdeI and purified. NI0 and NIPST0 were transcribed and radioactively labeled at the 5′ terminal end using T4 polynucleotide kinase. These RNAs revealed a single band corresponding to the full-length transcript of 231 nt (lanes 1,2). In vitro transcribed and radioactively labeled RNAs were incubated with RNase V1 (V1, 0.01 U/μL, lanes 5,6), RNase A (A, 1 ng/μL, lanes 7,8), or RNase T1 (T1, 0.1 U/μL, lanes 9,10). For sequencing reactions, RNAs were denatured and digested using RNase T1 (S, 0.1 U/μL in lanes 3 and 4). A ladder of alkaline hydrolyzed NI0 RNA fragments is shown (H, lane 11). Black arrowheads indicate sites sensitive to RNase V1 that were used to constrain Mfold. Sites sensitive to RNase A and RNase T1, which were used to constrain Mfold, are indicated by gray and white arrowheads, respectively. Positions of G nucleotides and stem–loops I, II, and III are indicated at the left. The secondary structure of the NRF IRES module was derived by using RNase accessibility data to constrain the Zuker Mfold provided by Rensselaer Polytechnic Institute (Troy, NY; http://www.bioinfo.rpi.edu/applications/mfold/old/rna/) using the following parameters: 37°C, 1 M NaCl, 5% suboptimality, ΔG = −21.30 kcal/mol. For a better overview, predicted secondary structures of nt 611–653 in wt and mutant human NRF are redrawn. Stem–loops I and II are indicated. Nucleotide numbers correspond to human NRF 5′UTR with respect to nt A (654) of the AUG codon. Point mutations are indicated in lower-case letters. In NI0, nt 649–652 were changed from GCUU to UGCA to obtain NIPST0. (B) HeLa cells were transfected with the following dicistronic constructs: p0RLFL containing no intercistronic sequences; p0RL-NI0-FL containing wt human NRF IRES module (nt 507–653); p0RL-NIPST0-FL containing point mutations in human NRF IRES module (649T, 650G, 651C, 652A); p0RL-NICG0-FL containing additional point mutations (641G, 642C, 643A); or p0RL-NID10-FL lacking nt 637–652 (containing two additional nucleotides). Details of these constructs are schematically presented at the top. Additionally, p0RL-NI0-FL was co-transfected with siRNA against Renilla luciferase (siRL) or a control siRNA (siS). White bars indicate the relative Renilla luciferase activity of cells transfected with the dicistronic plasmids. The Renilla luciferase activity in cells transfected with p0RL-NI0-FL was set to 1 in each single experiment. Relative IRES activities were determined as described in the legend for Fig. 2B. Shaded bars indicate the IRES activity of cells transfected with the dicistronic plasmids. The IRES activity of cells transfected with p0RL-NI0-FL (13-fold) was set to 1 in each experiment. (C) Integrity of dicistronic mRNAs. Northern blot analysis was performed with 10 μg of total RNA isolated from transfected HeLa cells. The RNA content was visualized by ethidium bromide (EtBr) staining (left). Human 28S (5.3 kb) and 18S RNA (2.3 kb) bands are indicated. RNA blot was hybridized with radioactively labeled full-length Renilla luciferase coding region (middle) or firefly luciferase coding region (right). RNA blot was then subjected to autoradiography. Transfected constructs are indicated above each lane. Dicistronic RNAs are indicated by the asterisk.
FIGURE 4.

Specific binding of cellular proteins to human NRF IRES module. Schematic diagram of the constructs used to generate wild-type (wt) (NI0) or mutant (NIPST0) of the NRF IRES module are shown at the top. Constructs were digested with XhoI and purified. The human NRF IRES module (NI0) or unspecific antisense control RNA (control RNA) was transcribed in vitro and coupled to magnetic beads by the poly(A) tail (AAA). Extracts from HeLa cells were purified using magnetic beads. After three wash steps (W1, W2, and W3), binding proteins were eluted by 0.5, 1.0, or 2 M KCl buffer. Wash and eluted fractions together with protein size marker were analyzed on a 10% denaturing SDS–polyacrylamide gel. Protein bands were detected by Coomassie blue staining. Protein marker band sizes are indicated at the left. The unspecific 45-kDa protein is indicated by asterisks. The specific 38-kDa protein is indicated by an arrow. This band was isolated and further analyzed by ESI/MS/MS.
We introduced NIPST0 and an additional mutation lacking nt 637–650 (NID10) into the human IRES module to investigate the role of a predicted stem–loop I in internal translation initiation. To isolate the effect of point mutations, we removed all nonessential linker sequences from pRLFL cistrons, thereby creating p0RLFL (Fig. 3B). Wild-type and mutant IRES modules were inserted into the intercistronic region of p0RLFL and then transfected into HeLa cells. Similar levels of Renilla luciferase were detected in all transfectants. To estimate the IRES activity, firefly luciferase activity was normalized to Renilla luciferase activity. For better comparison, the relative IRES activity of p0RL-NI0-FL (13-fold) was set to 1. As expected, complete deletion (p0RL-NID10-FL) or introduction of mutations that destroy the secondary structure of stem–loop I (p0RL-NIPST0-FL) significantly decreases relative IRES activity. Reconstruction of the stem–loop structure by introduction of two complementary bases (p0RL-NICG0-FL) significantly improves the relative IRES activity. Together, these data demonstrate the importance of nt 640–653 in NRF 5′UTR for internal translation initiation (Fig. 3B). We utilized siRNA against Renilla luciferase to confirm that Renilla and firefly luciferase are both translated from dicistronic mRNAs. As shown in Figure 3B, co-transfection of siRNA against Renilla luciferase coding sequence (siRL) significantly reduces both Renilla and firefly luciferase activities. In contrast, a negative control siRNA (siS) has no significant effect on Renilla and firefly luciferase activities. Furthermore, the integrity and steady-state level of dicistronic mRNAs were confirmed by Northern blot analysis using probes containing either Renilla or firefly luciferase coding region (Fig. 3C).
Identification of JKTBP1 as an NRF IRES-binding protein
To isolate the cellular proteins interacting with NRF IRES, in vitro transcribed human NRF IRES module RNA and antisense control RNA were coupled to magnetic beads and incubated with HeLa cell extracts. After washing, RNA-bound proteins were eluted by increasing KCl concentration and analyzed by SDS-PAGE and Coomassie staining (Fig. 4). Although Coomassie staining is insensitive to the detection of small amounts of proteins, it is most appropriate for further analysis by peptide mass fingerprinting via electrospray ionization and tandem mass spectrometry (ESI/MS/MS). At lower concentrations of KCl, many cellular proteins were eluted. At higher concentrations of KCl (1 or 2 M), two proteins with apparent molecular mass of 45 and 38 kDa were detected. Of the two proteins, only the 38-kDa protein binds specifically to the NRF IRES module (arrow). The 45kDa protein binds to both NRF IRES and control RNAs, suggesting an unspecific affinity to RNA or beads (asterisks).
The 38 kDa protein was excised from the gel shown here (Fig. 4) and analyzed by ESI/MS/MS. The obtained masses were compared with those of proteins in the Swiss-Prot database. The analyzed peptide covered 25% of the human JKTBP1 protein sequence.
Mapping of JKTBP1-binding site in the NRF IRES module
The specificity of JKTBP1 binding to the NRF IRES module was confirmed by EMSA assay using the GST–JKTBP1 fusion protein (Fig. 5A). The NRF IRES module alone or incubated with GST protein solely forms no complex (negative control, Fig. 5A, lanes 1,2). Notably, JKTBP1 shows indistinguishable binding affinity to wild-type (NI0) and mutant (NIPST0, lacking stem–loop I) NRF IRES modules (Fig. 5A, lanes 3,7). This indicates that JKTBP1 binding is independent of the predicted stem–loop I. A polyclonal anti-JKTBP1 antibody was established previously for Western blot applications (Akagi et al. 2000). This antibody inhibits binding of GST–JKTBP1 to the NRF IRES (Fig. 5A, lanes 4,8). Thus, we assume that binding of the polyclonal anti-JKTBP1 antibody interferes with the RNA-binding affinity of the JKTBP1 protein in vitro. Additional competition experiments demonstrate the specificity of JKTBP1 binding to NRF IRES. Formation of the JKTBP1 complex is significantly impaired by addition of specific cold competitor NRF IRES RNA but not by addition of nonspecific competitor transferrin receptor RNA (Fig. 5A, lanes 11–14). We note that cold competitors NI0 and NIPST0 equally impaired formation of the JKTBP1 complex (data not shown).
FIGURE 5.

JKTBP1 binds specifically to NRF IRES. GST or GST–JKTBP1 proteins were expressed in bacteria and purified. (A) Schematic diagram of the constructs used to generate transferrin receptor RNA (nonspecific cold competitor), wild-type (WT) (NI0), or mutant (NIPST0) of the NRF IRES module are shown at the top. Constructs were digested with NdeI or BamHI as indicated. GST or GST–JKTBP1 protein (1 μg) was incubated with in vitro transcribed human NRF IRES WT module NI0 or mutant module NIPST0 (lacking stem–loop I) in the presence or absence of nonlabeled NI0 transcript (specific cold competitor) or in vitro transcribed transferrin receptor RNA (nonspecific cold competitor). Rabbit polyclonal antibody against JKTBP1 (1 μg) was added to the reactions where indicated. (B) Schematic diagram of the constructs used to generate wt (NI0) or mutant (NIH0, NIS0) of NRF IRES module are shown at the top. Constructs were digested with NdeI or RsaI as indicated. GST or GST–JKTBP1 protein (1 μg) was incubated with in vitro transcribed human NRF IRES wt module NI0 or mutant module NIH0 and NIS0. Complexes were analyzed on native 6% polyacrylamide gel and then subjected to autoradiography. JKTBP1 complex and unbound RNA are indicated at the left.
Next, we used the HindIII restriction site at nt 579 to delete nt 579–636 spanning stem-loops II and III (NIH0). Equal amounts of NRF IRES module (NI0) and HindIII deletion mutant (NIH0) were incubated with GST or GST–JKTBP1 (Fig. 5B). Both GST and GST–JKTBP fail to bind to NIH0, indicating that binding of JKTBP1 to the NRF IRES module requires nt 579–636. Indeed, GST–JKTBP1 is able to bind to nt 579–639, indicating that this region contains a functional binding site for JKTBP1 protein (Fig. 5B, lane 9). The free RNA probe nt 579–639 forms various slower migrating bands in presence or absence of GST or GST–JKTBP1 (Fig. 5B, lanes 7–9). This is most probably due to formation of various secondary structures of the free RNA probe nt 579–639.
JKTBP1 and stem–loop I mediate NRF IRES module activity
To investigate the role of JKTBP1 in NRF IRES-dependent translation, we cotransfected HeLa cells with p0RL-NI0-FL, p0RL-NIPST0-FL, p0RLFL, or p0RL-NIH0-FL and increasing amounts of JKTBP1 expressing plasmid pJKTBP1. Western blot analysis using anti-JKTBP1 antibody shows that JKTBP1 is abundantly expressed in HeLa cells (Fig. 6A, pcDNA3) and that the expression level of JKTBP1 protein correlates with the amounts of transfected pJKTBP (Fig. 6A). Similarly, the endogenous amount of NRF protein was slightly elevated in JKTBP transfected cells. Western blot analysis using anti-p65 antibody served as internal control.
FIGURE 6.

Overexpression of JKTBP1 enhances NRF IRES activity. Dicistronic constructs p0RL-NI0-FL (containing wild-type [WT] human NRF IRES module), p0RL-NIPST0-FL (lacking stem–loop I), or p0RLFL (containing no intercistronic sequences) were transfected into HeLa cells together with increasing amounts of JKTBP1 expressing plasmid or an empty expressing control vector pcDNA3. (A) Transfected cells were subjected to Western blot analysis with rabbit polyclonal antibody against JKTBP1 or NRF or mouse monoclonal antibody against NF-κB p65 as control. At the top, results are representative for three independent experiments. JKTBP1, NRF, and p65 protein bands are indicated. Densitometric analysis of detected bands is shown ±SEM from three independent experiments. JKTBP bands were quantified and normalized to corresponding p65 signals in each experiment. (B) Relative Renilla luciferase activity of cells transfected with the dicistronic plasmids (white bars) is shown on the secondary axis. Renilla luciferase activity in cells transfected with p0RL-NI0-FL and pcDNA3 was set to 1 in each single experiment. The relative IRES activity is shown as the mean ratio of firefly and Renilla luciferase activities ±SEM from three independent experiments (black bars) on primary axis. The data were determined as described in the legend to Fig. 2B. IRES activity of cells transfected with p0RL-NI0-FL and pcDNA3 was set to 1 in each experiment. (C) Northern blot analysis was performed with 10 μg of total RNA isolated from transfected HeLa cells. RNA content was visualized by ethidium bromide (EtBr) staining (left). Human 28S (5.3 kb) and 18S RNA (2.3 kb) bands are indicated. RNA blot was hybridized with radioactively labeled full-length firefly luciferase coding region (right). RNA blot was then subjected to autoradiography. Transfected constructs are indicated above each lane. Dicistronic RNA is indicated by the asterisk. (D) The dicistronic construct p0RL-polio-FL containing polio IRES was transfected into HeLa cells together with increasing amounts of JKTBP1 expressing plasmid or an empty expression control vector pcDNA3. IRES activity of p0RL-polio-FL alone was set to 1 (data not shown).
The results of dicistronic assays show that overexpression of JKTBP1 augments the activity of the NRF IRES module in a dose-dependent manner (Fig. 6B, p0RL-NI0-FL). Interestingly, residual IRES activity of p0RL-NIPST0-FL is elevated by over-expression of JKTBP1. In contrast, JKTBP1 shows no or only minimal effect on translation of the downstream cistron in p0RL-NIH0-FL (Fig. 6B). Notably, there is almost no expression of firefly luciferase and thus no IRES activity detectable in p0RLFL or p0RL-NIH0-FL transfected cells (factor 0.035). Similar levels of Renilla luciferase were detected in all transfectants, and the level and integrity of dicistronic mRNA were confirmed by Northern blot analysis (Fig. 6C). To confirm that the interaction of JKTBP1 with NRF IRES is specific, we tested the effect of JKTBP1 on polio IRES-dependent translation. Dicistronic plasmid p0RL-polio-FL containing polio IRES was cotransfected with pJKTBP1 into HeLa cells. As shown in Figure 6D, JKTBP1 has no enhancing effect on polio IRES-dependent translation of the downstream cistron.
To further analyze the role of JKTBP1 in NRF IRES-dependent translation, we utilized mRNA knockdown using siRNA. Two different siRNAs against JKTBP1 (siRNA2 and siRNA3) and a scramble control RNA (scramble) were transfected together with p0RL-NI0-FL into HeLa cells. As shown in Figure 7A, endogenous JKTBP1 and NRF levels were reduced in siRNA-transfected cells compared with nontransfected cells or cells transfected with scramble siRNA. Compared to siRNA3, siRNA2 is more capable of reducing JKTBP1 and NRF protein levels in cells. Simultaneously, the IRES activity of p0RL-NI0-FL was reduced to 53.8% in siRNA2-transfected and to 66.4% in siRNA3-transfected cells. This reduction correlates with the capability of siRNA2 or siRNA3 in reduction of JKTBP1 protein level in cells. Notably, the endogenous level of JKTBP1 protein was significantly reduced but not completely abolished by siRNA2. This raises the question whether remaining IRES activity by siRNA2 is due to residual levels of JKTBP1 protein or the presence of nt 640–653 (stem–loop I). In order to address this question, we compared the relative factor of IRES activity in the presence or absence of JKTBP1 and stem–loop I (Fig. 7C). The data show that reduction of JKTBP1 has a lower inhibitory effect (53.8%) on relative IRES module activity compared to functional mutations in stem–loop I (17.9%). Most importantly, simultaneous reduction of JKTBP1 and mutation of stem–loop I substantially diminish NRF IRES module activity to 0.9%. Thus, the remaining IRES activity observed by siRNA2 (Fig. 7A) is most likely due to presence of stem–loop I. Furthermore, these results clearly demonstrate that binding of JKTBP1 and the 14 nt element both together are crucial for NRF IRES module activity.
FIGURE 7.

Endogenous JKTBP1 protein augments the activity of NRF IRES module. HeLa cells were transfected with p0RL-NI0-FL (control) or with both p0RL-NI0-FL and JKTBP1 siRNAs (siRNA2 and siRNA3) and control siRNA (scramble). (A) At 48 h after transfection, extracts of transfected cells were prepared and subjected to Western blot analysis with rabbit polyclonal antibodies against JKTBP1 or NRF peptide 25–45 or rabbit polyclonal antibody against NF-κB p65 as control. JKTBP1 and p65 protein bands are indicated. (B) At 48 h after transfection, extracts of transfected cells were subjected to Dual-Luciferase activity assay (Promega). IRES activity was determined as described in the legend to Figure 2B. Renilla luciferase activity of transfected cells (white bars) is shown on the secondary axis. Renilla luciferase activity in cells transfected with p0RL-NI0-FL and pcDNA3 was set to 1 in each experiment. Relative IRES activity is shown as the mean ratio of firefly and Renilla luciferase activities ±SEM from three independent experiments (black bars) on the primary axis. IRES activity of cells transfected with p0RL-NI0-FL (control) was set to 100% in each experiment. (C) HeLa cells were transfected with indicated dicistronic plasmids and siRNA2 against JKTBP1 where indicated. At 48 h after transfection, extracts of transfected cells were subjected to Dual-Luciferase activity assay (Promega). IRES activity was determined as described in the legend to Figure 2B. Relative IRES activity is shown as the mean ratio of firefly and Renilla luciferase activities ±SEM from three independent experiments (black bars). IRES activity of cells transfected with p0RFL-NI0-FL was set to 100% in a single experiment.
DISCUSSION
Since the first evidence for internal translation initiation in cellular mRNAs (Macejak and Sarnow 1991), dissection of cellular IRES modules has been considered the most common approach to identify functional elements involved in internal translation initiation. In this study, we used sequence homology between human and murine NRF 5′-UTRs as a guiding factor in determining a minimal functional IRES module. IRES activity of the homologous 150 nt modules was proven in dicistronic constructs. This dicistronic assay is considered a valid method to compare IRES activity of different constructs as complemented by proof of equal expression of first cistron and integrity of dicistronic mRNA. More precisely, several important control experiments were performed to validate the integrity of dicistronic mRNAs and to exclude the presence of cryptic promoters or aberrant splicing that could lead to IRES-independent translation of the downstream cistron. First, a specific probe hybridizing with the downstream cistron exclusively identified full-length dicistronic mRNAs in Northern blot analysis (Fig. 3C, 6C). Second, promoterless dicistronic constructs revealed negligible activity of the downstream cistron (Fig. 2C). Third, transfection of siRNA against the upstream cistron significantly decreases activity of the downstream cistron (Fig. 3B).
Initially, successive 3′ deletion of human and murine NRF 5′-UTRs in dicistronic constructs suggested that 50 nt immediately upstream of the NRF start codon (nt 600–653 of human or nt 800–857 of murine NRF 5′-UTR) are required for NRF IRES activity (Fig. 2B). Secondary-structure analysis of the NRF 5′-UTR revealed a small stem–loop close to the AUG start codon (nt 640–653, stem–loop I). Point mutations altering formation of this stem–loop substantially diminished the IRES activity (Fig. 3B, p0RL-NIPST0-FL). Thus, we suggest that stem–loop I forms an essential structural element of IRES function (Fig. 3B, p0RL-NID10-FL). Interestingly, point mutations that interfere with the structure of stem–loop I caused no apparent effect on binding of JKTBP1 (Fig. 5A) or crude cellular proteins to the IRES module (data not shown). Similarly, deletion of stem–loop I showed no effect on binding of JKTBP1 or cellular proteins (NID10, data not shown). Despite extensive efforts, stem–loop 651 (nt 40–653) alone failed to form any complexes with cellular proteins (data not shown). Although this might be due to technical difficulties in creation or detection of a 14-nt stem–loop complex, it is possible that stem–loop I is exclusively a structural element independent of cellular proteins. In viral IRES modules, tRNA-homologous sequences were found to drive internal entry of ribosomes (Lyons and Robertson 2003). In the 5′-UTR of the homeodomain protein Gtx, a 9 nt IRES was shown to be 100% complementary to 18S rRNA, suggesting that base pairing between the 9 nt segment and 18S rRNA can lead to ribosome recruitment (Chappell et al. 2000, 2004). The NRF IRES module does not show significant homology with 18S rRNA or tRNA sequences. Still, our observations suggest that the structure of nt 640–653 contributes to internal initiation of translation.
Very little is known about cellular function of JKTBP1. Alignment of the JKTBP1 protein sequence revealed two adjacent RNA binding domains (RBDs) closely related to hnRNPs (Akagi et al. 2000). Many hnRNPs contain multiple RBDs, which can bind to more than one RNA element simultaneously (Kamei et al. 1999; Bandiera et al. 2005). Here, several observations using the GST–JKTBP1 fusion protein show that JKTBP1 specifically binds to nt 578–639 in the NRF IRES module (Fig. 5B). Previously, JKTBP1 was shown to bind to AU-rich RNA elements in human TNF-α and COX-2 mRNAs (Dean et al. 2002). However, comparison of the NRF IRES module sequence with TNF-α and COX-2 mRNAs revealed no significant homology. Previously, possible binding sites of JKTBP1 were identified from pools of 20 nt random sequence RNA in vitro (Kamei and Yamada 2002). A JKTBP1 consensus high-affinity site was found to be GGACUAGCC, showing no significant homology with the JKTBP1-binding region in the NRF IRES module (nt 578–639). Thus, it is conceivable that the bordering primary or secondary structure of the NRF IRES module can affect JKTBP1-binding specificity.
Our data provide evidence that JKTBP1 is able to augment activity of the NRF IRES module in dicistronic constructs. Previous studies have shown that JKTBP1 shuttles between the nucleus and cytoplasm (Kawamura et al. 2002). Cytoplasmic accumulation of the JKTBP–RNA complex was supposed to increase stability of RNA or decrease nuclear re-import of RNA (Suzuki et al. 2005). In this study, however, over-expression of JKTBP1 has no significant effect on expression of the first cistron or the entire level of dicistronic mRNAs (Fig. 6B,C). Furthermore, JKTBP1 shows no effect on polio IRES under identical experimental circumstances (e.g., vector sequences, DNA concentration). These data also emphasize that JKTBP1 acts directly on the NRF IRES module.
In agreement with results from dicistronic assays, JKTBP1 elevates endogenous NRF protein level in cells (Fig. 6A). Accordingly, the endogenous NRF protein level was reduced by down-regulation of JKTBP1 (Fig. 7A). These results together with those from dicistronic assays strongly suggest that JKTBP1 contributes to IRES-mediated translation of endogenous NRF mRNAs. Certainly, however, the total amount of endogenous NRF protein is most likely regulated at several levels. In particular, endogenous NRF protein is encoded by two mRNAs bearing two different 3′-UTRs. Previous studies have shown that expression of these mRNAs is not equal in cells, suggesting a regulatory role of 3′-UTRs (Nourbakhsh and Hauser 1999). In this study, we mainly focused on translational activity of an isolated NRF IRES module. However, NRF 5′-UTR apparently contains functional upstream elements in addition to the IRES module. For instance, deletion of nt 001–500 (pRL501–653FL) reduces IRES activity to ∼72% compared to full-length NRF 5′-UTR (Fig. 2B, pRL001–653FL,). In summary, these observations suggest that the IRES module, in concert with upstream elements and 3′-UTRs, determine the total amount of endogenous NRF protein in cells.
Our data show that JKTBP1 and the 14-nt element forming stem–loop I both together essentially contribute to NRF IRES module activity (Fig. 7C). However, several observations suggest that JKTBP1 and the 14-nt element close to AUG start codon act independently. As mentioned above, EMSA experiments indicate that binding of JKTBP1 to the IRES module does not depend on the 14-nt element. Accordingly, siRNA2 against JKTBP1 reduces NRF IRES module activity by the presence or absence of functional mutations within stem–loop I (Fig. 7C, p0RL-NIPST0-FL+siRNA2). By comparison, point mutations within stem–loop I show a more significant effect on NRF IRES activity (Fig. 7C, p0RL-NIPST0-FL, 17.9%) than siRNA2 (Fig. 7C, p0RL-NI0-FL+siRNA2, 53.8%). Nevertheless, the results of this study clearly demonstrate that JKTBP1 and the 14-nt element are two discretely acting components of the NRF 5′-UTR, which are required for full activity of NRF IRES module.
Two studies suggested a possible role of JKTBP1 in regulation of cytokine gene expression. JKTBP1 was shown to bind to TNF-α 3′-UTR (Dean et al. 2002). By proteomic analysis, the JKTBP1 protein level was found to be elevated by IL-1 stimulation in primary epithelial cells (Barcelo-Batllori et al. 2002). Consistent with a possible role of JKTBP1 in regulation of cytokine gene expression, our study indicates that JKTBP1 augments endogenous expression of NRF protein regulating cytokine genes in HeLa cells (Nourbakhsh et al. 2001). In contrast to primary cells, the expression pattern of JKTBP1 protein was not changed upon IL-1 stimulation in HeLa cells (data not shown). Nevertheless, the amount of JKTBP1 significantly modulates NRF expression in HeLa cells (Fig. 6A, 7A). Thus, we presume a general regulatory link between JKTBP1, NRF, and cytokine expression, which needs to be confirmed by additional experiments in primary epithelial cells.
MATERIALS AND METHODS
Plasmid construction
pRLFL is a derivative of the expression plasmid pSBC-1 (accession number X68257) and was previously described as pBSRF (Oumard et al. 2000). The NRF 5′-UTR sequences were amplified by PCR and inserted into pRLFL between the stop codon of Renilla luciferase and the AUG codon of firefly luciferase. pFL was created by deletion of PmeI/SacI fragment in p0RL-NI0-FL containing Renilla coding sequence and NRF 5′UTR sequences. Reporter plasmids DRL001–653FL and DRLmu001–807FL were created by deletion of SV40 promoter region (ClaI/XhoI fragments) in pRL001–653FL and pRLmu001–807FL, respectively. D001–653FL and Dmu001–807FL were created by deletion of SV40 promoter region and Renilla luciferase coding sequences (ClaI/XbaI fragments) in pRL001–653FL and pRLmu 001–807FL, respectively. Chloramphenicol acetyltransferase (CAT) expression plasmid, pMPSV-HECAT, encodes bacterial CAT and was described earlier (Artelt et al. 1988). p0RLFL was created by deletion of ∼50 base pairs (bp) of linker sequences of pRLFL located between XhoI and PfoI sites downstream from the transcription start site. Mutagenesis of IRES sequences was performed using the QuikChange site-directed mutagenesis kit (Stratagene). Plasmids for in vitro transcription were constructed using amplified PCR fragments containing desired NRF IRES sequences cloned to EcoRI and XhoI of pBluescript SK+ (Stratagene, accession number X52327). JKTBP1 expression plasmid (pJKTBP) was constructed by cloning of the EcoRI/SmaI fragment of pEGFPC1-JKTBP1 (Kawamura et al. 2002) into EcoRI/EcoRV sites of pcDNA3.1 (Invitrogen). Dicistronic construct p0RL-polio-FL containing polio IRES is a derivative of the expression plasmid pSBC-1 (Dirks et al. 1993) and was created by deletion of linker sequences between the transcription start site and Renilla luciferase coding sequences in pBSRPF, as described earlier (Oumard et al. 2000).
Cell lines and DNA transfection
HeLa or C243 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (BIO-Whittaker), 2 mM glutamine, penicillin, and streptomycin and were transfected by calcium phosphate co-precipitation. A total of 5 μg DNA containing 1 μg of reporter plasmid, 1 μg of CAT control plasmid (where indicated), and a suitable complementing amount of high molecular weight DNA was transfected per 7.5 × 104 cells. Cells were harvested after 72 h and pooled for reporter gene assays.
siRNA transfection
HeLa cells were transfected by calcium phosphate co-precipitation. A total of 5 μg DNA containing 2.5 μg of reporter plasmid and 113 nM siRNAs was transfected per 7.5 × 104 cells. Cells were harvested after 48 h and pooled for reporter gene assays.
JKTBP1-siRNAs
siRNA2:
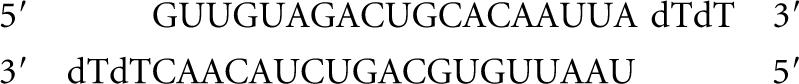 |
siRNA3:
Scramble:
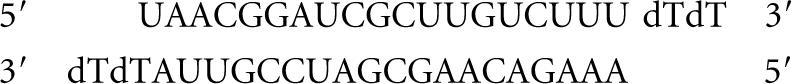 |
Renilla luciferase-siRNAs
siRL:
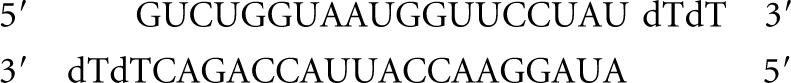 |
siS:
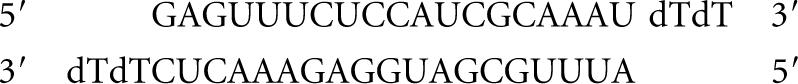 |
Reporter gene assay
Firefly and Renilla luciferase activities were determined using the Dual-Luciferase reporter system (Promega) as described in the manufacturer's protocol. The amount of protein was determined using BCA reagent (Sigma-Aldrich) as described in the manufacturer's protocol. The amount of CAT enzyme was measured with the CAT-ELISA kit (Boehringer) according to the manufacturer's protocol. Luciferase activity was measured in a Lumat LB 9501 luminometer (Berthold), reported as relative light units (RLUs), and corrected for protein content and the amount of CAT enzyme as a transfection efficiency control.
Northern blot analysis
Total RNA from cells was isolated using TRIZOL reagent (Gibco/BRL, Life Technologies) according to the manufacturer's instructions. Poly(A)+ RNA was isolated from total RNA using Dynabeads Oligo(dT)25 (Dynal) according to the manufacturer's instructions. Northern blot analysis was performed as described earlier (Oumard et al. 2000). Firefly and Renilla luciferase specific fragments were obtained by digestion of p0RL-NI0-FL with XbaI and EcoRV and phRL-TK (Promega) with NheI and XbaI enzymes, respectively. The firefly luciferase XbaI/EcoRV fragment and Renilla luciferase XbaI/NheI fragment were analyzed and isolated from 1% agarose gel containing coding sequences of firefly luciferase (aa 17–447) and coding sequences of Renilla luciferase (aa 1–311), respectively. NRF- and actin-specific probes were described earlier containing coding sequences of human NRF (aa 18–398) or β-actin cDNA sequence (1–1793), respectively (Nourbakhsh and Hauser 1999; Nourbakhsh et al. 2001).
Western blot analysis
Cellular extracts were prepared as described in the Dual-Luciferase assay protocol; 20 μg of extracts were loaded onto a 10% polyacrylamide gel. Western blot analysis was performed as described earlier (Oumard et al. 2000) using 1:500 diluted rabbit polyclonal antibodies against GST–JKTBP1 (Kamei et al. 1999; Akagi et al. 2000) or NRF peptide 25–45 (Nourbakhsh and Hauser 1999). Monoclonal antibody against p65 (sc8008) and polyclonal goat antibodies against eIF4G (sc9601) or eIF4GII (sc13787) were obtained from Santa Cruz Biotechnology, Inc.
In vitro transcription
Templates for in vitro transcription were prepared by linearization of pBluescript SK+ plasmids (Stratagene) with restriction enzymes as indicated in Figures 4 and 5, A and B. In vitro transcription reactions with the purified linearized templates were carried out with the T7-Riboprobe kit (Promega) using unlabeled or radioactively labeled rUTP (as indicated) following the manufacturer's instructions.
RNA sequencing and structure analysis
RNAs (NI0 and NIPST0) were transcribed in vitro using the T7-Riboprobe kit (Promega). The 5′ ends of the transcripts were labeled using the KinaseMax kit (Ambion). RNA sequencing and structure analysis were performed using RNA-grade nuclease kits (Ambion) according to the manufacturer's protocols.
Preparation of cell extracts
Nuclear and cytoplasmic extracts were prepared as described earlier (Schreiber et al. 1989).
Expression and purification of GST fusion proteins
Recombinant proteins were expressed and purified as fusion proteins with GST in Escherichia coli BL21 cells as described earlier (Tsuchiya et al. 1998; Suzuki et al. 2005).
RNA-shift analysis
In vitro synthesized radioactive transcript (5 × 105 cpm) was incubated with 30 μg of HeLa cytoplasmic extract or 1 μg of purified GST or GST–JKTBP proteins in 20 mM HEPES, pH 7.9, 10 mM KCl, 1 mM CaCl2, 2 mM MgCl2, 2 μg of t-RNA competitor from E. coli (Roche), and 2% Ficoll for 10 min at 37°C. RNAse T1 (30 U, Roche) was added to the mixture and incubated for 30 min at 37°C. The complexes were then analyzed by 6% native polyacrylamide gel electrophoresis and autoradiography of the dried gel.
Purification and sequencing of IRES-binding proteins
Oligo-dT beads (100 μL, Dynal Biotech) were equilibrated with binding buffer (20 mM Tris-HCl, pH 7.5, 150 mM KCl, 2 mM EDTA) and loaded with ∼5 μg of in vitro synthesized polyadenylated IRES RNA; 2.5 mg of cytoplasmic extract was then added to the RNA-loaded beads, which were then incubated for 15 min at 30°C. The beads were then washed three times in 20 mM HEPES, pH 7.5, 150 mM KCl, 2 mM MgCl2, 0.5 mM DTT, 0.5 mM PMSF, 5% glycerol, and 0.1% NP-40. Binding proteins were eluted with increasing KCl concentrations (0.5, 1, and 2 M) in washing buffer. After SDS sample buffer was added, samples were incubated at 95°C for 10 min and analyzed by SDS–polyacrylamide gel electrophoresis, which was followed by Coomassie staining. Detected protein bands were excised from the gel and washed twice with water. After digestion with trypsin, peptides were purified by nanocapillary HPLC and sequences were reconstructed by ESI/MS/MS (Eurogentec). Sequence information was then compared to the MRC-Mass Spectrometry Protein Sequence DataBase for identification.
ACKNOWLEDGMENTS
We thank U. Dreikhausen (Hannover Medical School) for discussions and critical reading of the manuscript. We are grateful for financial support through a HiLF grant from Hannover Medical School and a Ph.D. grant from KAAD.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.545407.
REFERENCES
- Akagi, T., Kamei, D., Tsuchiya, N., Nishina, Y., Horiguchi, H., Matsui, M., Kamma, H., Yamada, M. Molecular characterization of a mouse heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its tissue-specific expression. Gene. 2000;245:267–273. doi: 10.1016/s0378-1119(00)00032-9. [DOI] [PubMed] [Google Scholar]
- Artelt, P., Morelle, C., Ausmeier, M., Fitzek, M., Hauser, H. Vectors for efficient expression in mammalian fibroblastoid, myeloid and lymphoid cells via transfection or infection. Gene. 1988;68:213–219. doi: 10.1016/0378-1119(88)90023-6. [DOI] [PubMed] [Google Scholar]
- Bandiera, A., Medic, N., Akindahunsi, A.A., Manzini, G. In vitro dual binding activity of a evolutionarily related subgroup of hnRNP proteins. Mol. Cell. Biochem. 2005;268:121–127. doi: 10.1007/s11010-005-3700-1. [DOI] [PubMed] [Google Scholar]
- Barcelo-Batllori, S., Andre, M., Servis, C., Levy, N., Takikawa, O., Michetti, P., Reymond, M., Felley-Bosco, E. Proteomic analysis of cytokine induced proteins in human intestinal epithelial cells: Implications for inflammatory bowel diseases. Proteomics. 2002;2:551–560. doi: 10.1002/1615-9861(200205)2:5<551::AID-PROT551>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Byrd, M.P., Zamora, M., Lloyd, R.E. Translation of eukaryotic translation initiation factor 4GI (eIF4GI) proceeds from multiple mRNAs containing a novel cap-dependent internal ribosome entry site (IRES) that is active during poliovirus infection. J. Biol. Chem. 2005;280:18610–18622. doi: 10.1074/jbc.M414014200. [DOI] [PubMed] [Google Scholar]
- Chappell, S.A., Edelman, G.M., Mauro, V.P. A 9-nt segment of a cellular mRNA can function as an internal ribosome entry site (IRES) and when present in linked multiple copies greatly enhances IRES activity. Proc. Natl. Acad. Sci. 2000;97:1536–1541. doi: 10.1073/pnas.97.4.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell, S.A., Edelman, G.M., Mauro, V.P. Biochemical and functional analysis of a 9-nt RNA sequence that affects translation efficiency in eukaryotic cells. Proc. Natl. Acad. Sci. 2004;101:9590–9594. doi: 10.1073/pnas.0308759101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean, J.L., Sully, G., Wait, R., Rawlinson, L., Clark, A.R., Saklatvala, J. Identification of a novel AU-rich-element-binding protein which is related to AUF1. Biochem. J. 2002;366:709–719. doi: 10.1042/BJ20020402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks, W., Wirth, M., Hauser, H. Dicistronic transcription units for gene expression in mammalian cells. Gene. 1993;128:247–249. doi: 10.1016/0378-1119(93)90569-o. [DOI] [PubMed] [Google Scholar]
- Feng, X., Guo, Z., Nourbakhsh, M., Hauser, H., Ganster, R., Shao, L., Geller, D.A. Identification of a negative response element in the human inducible nitric oxide synthase (hiNOS) promoter: The role of NF-κB repressing factor (NRF) in basal repression of the hiNOS gene. Proc. Natl. Acad. Sci. 2002;99:14212–14217. doi: 10.1073/pnas.212306199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froese, N., Schwarzer, M., Niedick, I., Frischmann, U., Koster, M., Kroger, A., Mueller, P.P., Nourbakhsh, M., Pasche, B., Reimann, J., et al. Innate immune responses in NF-κB-repressing factor-deficient mice. Mol. Cell. Biol. 2006;26:293–302. doi: 10.1128/MCB.26.1.293-302.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen, C.U., Sarnow, P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes & Dev. 2001;15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- Kamei, D., Yamada, M. Interactions of heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its domains with high-affinity binding sites. Gene. 2002;298:49–57. doi: 10.1016/s0378-1119(02)00926-5. [DOI] [PubMed] [Google Scholar]
- Kamei, D., Tsuchiya, N., Yamazaki, M., Meguro, H., Yamada, M. Two forms of expression and genomic structure of the human heterogeneous nuclear ribonucleoprotein D-like JKTBP gene (HNRPDL) Gene. 1999;228:13–22. doi: 10.1016/s0378-1119(99)00020-7. [DOI] [PubMed] [Google Scholar]
- Kawamura, H., Tomozoe, Y., Akagi, T., Kamei, D., Ochiai, M., Yamada, M. Identification of the nucleocytoplasmic shuttling sequence of heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its interaction with mRNA. J. Biol. Chem. 2002;277:2732–2739. doi: 10.1074/jbc.M108477200. [DOI] [PubMed] [Google Scholar]
- Komar, A.A., Hatzoglou, M. Internal ribosome entry sites in cellular mRNAs: Mystery of their existence. J. Biol. Chem. 2005;280:23425–23428. doi: 10.1074/jbc.R400041200. [DOI] [PubMed] [Google Scholar]
- Lyons, A.J., Robertson, H.D. Detection of tRNA-like structure through RNase P cleavage of viral internal ribosome entry site RNAs near the AUG start triplet. J. Biol. Chem. 2003;278:26844–26850. doi: 10.1074/jbc.M304052200. [DOI] [PubMed] [Google Scholar]
- Macejak, D.G., Sarnow, P. Internal initiation of translation mediated by the 5′ leader of a cellular mRNA. Nature. 1991;353:90–94. doi: 10.1038/353090a0. [DOI] [PubMed] [Google Scholar]
- Mountford, P., Zevnik, B., Duwel, A., Nichols, J., Li, M., Dani, C., Robertson, M., Chambers, I., Smith, A. Dicistronic targeting constructs: reporters and modifiers of mammalian gene expression. Proc. Natl. Acad. Sci. 1994;91:4303–4307. doi: 10.1073/pnas.91.10.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourbakhsh, M., Hauser, H. Constitutive silencing of IFN-β promoter is mediated by NRF (NF-κB-repressing factor), a nuclear inhibitor of NF-κB. EMBO J. 1999;18:6415–6425. doi: 10.1093/emboj/18.22.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourbakhsh, M., Kalble, S., Dorrie, A., Hauser, H., Resch, K., Kracht, M. The NF-κb repressing factor is involved in basal repression and interleukin (IL)-1-induced activation of IL-8 transcription by binding to a conserved NF-κb-flanking sequence element. J. Biol. Chem. 2001;276:4501–4508. doi: 10.1074/jbc.M007532200. [DOI] [PubMed] [Google Scholar]
- Oumard, A., Hennecke, M., Hauser, H., Nourbakhsh, M. Translation of NRF mRNA is mediated by highly efficient internal ribosome entry. Mol. Cell. Biol. 2000;20:2755–2759. doi: 10.1128/mcb.20.8.2755-2759.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber, E., Matthias, P., Muller, M.M., Schaffner, W. Rapid detection of octamer binding proteins with ‘mini-extracts,’ prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, I., Itin, A., Einat, P., Skaliter, R., Grossman, Z., Keshet, E. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: Implications for translation under hypoxia. Mol. Cell. Biol. 1998;18:3112–3119. doi: 10.1128/mcb.18.6.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M., Iijima, M., Nishimura, A., Tomozoe, Y., Kamei, D., Yamada, M. Two separate regions essential for nuclear import of the hnRNP D nucleocytoplasmic shuttling sequence. FEBS J. 2005;272:3975–3987. doi: 10.1111/j.1742-4658.2005.04820.x. [DOI] [PubMed] [Google Scholar]
- Tsuchiya, N., Kamei, D., Takano, A., Matsui, T., Yamada, M. Cloning and characterization of a cDNA encoding a novel heterogeneous nuclear ribonucleoprotein-like protein and its expression in myeloid leukemia cells. J. Biochem. 1998;123:499–507. doi: 10.1093/oxfordjournals.jbchem.a021964. [DOI] [PubMed] [Google Scholar]
- Vagner, S., Gensac, M.C., Maret, A., Bayard, F., Amalric, F., Prats, H., Prats, A.C. Alternative translation of human fibroblast growth factor 2 mRNA occurs by internal entry of ribosomes. Mol. Cell. Biol. 1995;15:35–44. doi: 10.1128/mcb.15.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q., Sarnow, P. Location of the internal ribosome entry site in the 5′ non-coding region of the immunoglobulin heavy-chain binding protein (BiP) mRNA: Evidence for specific RNA–protein interactions. Nucleic Acids Res. 1997;25:2800–2807. doi: 10.1093/nar/25.14.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]