

Abstract
Background
Treatment of Stimulant-Use Disorders remains a formidable challenge, and the dopamine transporter (DAT) remains a potential target for antagonist or agonist-like substitution therapies.
Methods
This review focuses on DAT ligands, such as benztropine, GBR 12909, modafinil, and DAT substrates derived from phenethylamine or cathinone that have atypical DAT-inhibitor effects, either in vitro or in vivo. The compounds are described from a molecular mechanistic, behavioral, and medicinal-chemical perspective.
Results
Possible mechanisms for atypicality at the molecular level can be deduced from the conformational cycle for substrate translocation. For each conformation, a crystal structure of a bacterial homolog is available, with a possible role of cholesterol, which is also present in the crystal of drosophila DAT. Although there is a direct relationship between behavioral potencies of most DAT inhibitors and their DAT affinities, a number of compounds bind to the DAT and inhibit dopamine uptake but do not share cocaine-like effects. Such atypical behavior, depending on the compound, may be related to slow DAT association, combined sigma-receptor actions, or bias for cytosol-facing DAT. Some structures are sterically small enough to serve as DAT substrates but large enough to also inhibit transport. Such compounds may display partial DA releasing effects, and may be combined with release or uptake inhibition at other monoamine transporters.
Conclusions
Mechanisms of atypical DAT inhibitors may serve as targets for the development of treatments for stimulant abuse. These mechanisms are novel and their further exploration may produce compounds with unique therapeutic potential as treatments for stimulant abuse.
Keywords: DAT Inhibitors, DA Releasers, Dopamine Transporter, Atypical Effects, Molecular Mechanisms, Stimulant Abuse Treatment Compounds
1. Introduction
Stimulant-Use Disorder is defined in the Diagnostic and Statistical Manual of Psychiatric Disorders, 5th edition (DSM-5) as a pattern of use of amphetamine-like compounds, cocaine, or other stimulants resulting in clinically significant impairment or distress as manifested by at least two out of eleven listed problems within a 12-month period (American Psychiatric Association, 2013). Prevalence at a national level is most frequently estimated from the National Survey on Drug Use and Health (NSDUH), an annual household-based survey among individuals at least 12 years of age in the US population (see Gerlach et al., 2014). Past-year non-medical use of prescription stimulants in 2012 was between 2 and 4% in different age categories between 16 and 34 years of age; comparable numbers were for illicit stimulants 1 – 4% and cocaine (including crack) 1 – 5% (NSDUH data available from Substance Abuse and Mental Health Services Administration (SAMSHA) website). In comparison, the “Monitoring the Future” survey (Johnston et al., 2014) in selected schools indicates for 12th grade students in 2012 a past-year non-medical use of 7.9% of amphetamines (including methamphetamine) and 2.9% of cocaine. The amount of non-medical use that leads to Stimulant-Use Disorders is unknown. In this context, it can be noted that stimulants accounted for 3.3% of all drug-related emergency department visits for non-medical drug use in 2011 (Gerlach et al., 2014). Trends in Admissions by Primary Substance of Abuse (TEDS) to drug abuse treatment facilities show that 6% of all admissions in 2011 were for primary involvement of methamphetamine/amphetamines and 8% for cocaine (including crack). In this respect, stimulants and cocaine (14%) are similar to marijuana/hashish (18%), following on the heels of opiates (25%) and alcohol (39%; TEDS data available from SAMSHA website). Statistics for 2011 on Poison Centers showed the involvement of stimulants and street drugs (excluding analgesics) in 3% of all exposures (Gerlach et al., 2014).
Treatment of Stimulant-Use Disorders remains a formidable challenge. In the absence of effective pharmacotherapy, behavioral therapies comprise the mainstay of treatment. Pharmacotherapeutic potential has been tested for numerous compounds. Among non-stimulant treatment agents tried, the following have moderate potential and warrant future studies: naltrexone, disulfiram for individuals with a certain genotype, doxazosin, and vaccines (for recent review see Phillips et al., 2014). Given that psychostimulants target the dopamine transporter (DAT), the DAT became an early preclinical focus for discovery of treatment compounds. A seminal study by Kitayama et al. (1992) identified DAT residues differentially important for cocaine binding and DA uptake, opening up the possibility to block the effect of cocaine with a compound that overlaps the binding domain of cocaine but not that of dopamine (Dopamine Sparing Cocaine Antagonist, see Rothman et al. (2002)). Until recently, such an antagonist has been elusive (see section 4 below).
Separate from the idea of a potential cocaine antagonist is the interest in DAT as a target for agonist-like substitution therapy (Grabowski et al., 2001; Negus and Mello, 2003; Grabowski et al., 2004; Negus et al., 2007; Mello and Negus, 2007); see also recent review by (Howell and Negus, 2014). There is promising pre-clinical and clinical evidence for efficacy of the DAT substrates d-amphetamine, d-phenmetrazine and sustained-release d-methamphetamine (Negus et al., 2009; Banks et al., 2013a; 2013c; Phillips et al., 2014), making a strong case for further work on this group of medications. Prodrug formulations of d-amphetamine (lisdexamfetamine) and phenmetrazine (phendimetrazine), for which abuse has not been reported, are especially promising. Oral cocaine in coca tea does not lead to misuse and has been argued to show promise as agonist substitution therapy (Llosa and Llosa, 2005). Furthermore, modafinil has potential efficacy in treating patients with cocaine use disorders. Modafinil blunted cocaine-induced euphoria in controlled human laboratory studies (Dackis et al., 2003; Malcolm et al., 2006; Hart et al., 2008). An early clinical trial pointed to an appreciable positive effect of modafinil on drug use outcome (Dackis et al., 2005), but other trials gave mixed results (Anderson et al., 2009; Dackis et al., 2012). As pointed out in a Commentary by O'Brien (2012), patients addicted to cocaine with additional alcohol dependence do not reduce their cocaine use with modafinil (see also Anderson et al., 2009). Modafinil appears most useful for the treatment of moderate cocaine use disorder in combination with behavioral therapy. (The material on modafinil was covered by the presentation by Charles O'Brien in the Behavior, Biology, and Chemistry: Translational Research in Addiction 2014 symposium. Due to other commitments, Dr. O'Brien could not participate in this review.) Modafinil shares properties with other compounds now recognized as “atypical” DAT inhibitors, and along with recently developed atypical DA releasers it renews interest in the DAT as a pharmacotherapeutic target for treatment of Stimulant-Use Disorders.
Atypical DAT ligands are those that have effects that deviate from those expected, either in vitro or in vivo (Tanda et al., 2009; Schmitt et al., 2013). Typical DAT blockers, at high enough concentrations or doses are expected to (i) to fully inhibit DA uptake, and (ii) to fully inhibit binding of another blocker, as well as release of substrate by reversed transport. Typical DAT releasers are expected to fully release another substrate accumulated in cell or synaptosomes. Behaviorally, typical DAT blockers or releasers are expected to (i) stimulate locomotor behavior, and (ii) reinforce behavior, and as a result be subject to abuse. Examples of typical DAT blockers or releasers are cocaine or amphetamine, respectively. Examples of atypical DAT inhibitors are benztropine (BZT) and GBR 12909 (for more details see reviews by Tanda et al. (2009) and Schmitt et al. (2013)). Examples of atypical DAT releasers are 3,4-methylenedioxyethylamphetamine (MDEA) and PAL-1045 (Rothman et al., 2005; 2012).
2. Dopamine Transporter: Looking Under the Hood for Atypicality at the Molecular Level
2.1. Conformational cycle for dopamine uptake
In order to understand possible mechanisms for atypicality at the molecular level, it is important to examine the conformational cycle for substrate translocation. Fig. 1A shows different conformational stages of the DAT during a DA uptake cycle, depicted for a homology model of hDAT based on the bacterial leucine transporter (LeuT), a prokaryotic member of the neurotransmitter/sodium symporter (NSS) protein family (Yamashita et al., 2005; Zhou et al., 2007; Singh et al., 2007; 2008; Zhou et al., 2009; Krishnamurthy and Gouaux, 2012; Wang et al., 2013). The following is a brief summary of what is presented in more detail in our previous review (Schmitt et al., 2013), complemented with structural information obtained from the crystal structure of the drosophila DAT (dDAT) that was recently published (Penmatsa et al., 2013). Evolutionarily, dDAT is a closer relative of hDAT than LeuT.
Figure 1.

(A) Model of the conformational cycle for substrate translocation by the dopamine transporter (DAT), based upon crystal structures of the bacterial NSS family protein LeuT. In its default ligand-free (apo) configuration, the transporter protein is thought to be in dynamic equilibrium between outward- and inward-facing conformational states (upper left and lower left structures, respectively). Binding of extracellular Na+ ions at the S1 site stabilizes an open-to-out conformation with a fully open extracellular gate (upper right structure), allowing substrate molecules maximum access to the core S1 binding domain. Substrate binding at the S1 site induces closure of the extracellular gate, establishing an occluded, closed-to-out conformation (lower right structure). It has been suggested that interaction of a second substrate molecule with a secondary binding domain in the extracellular vestibule (the S2 site, located 11-13 Å above the S1 site) helps facilitate opening of the intracellular gating network, giving rise to a fully inward-facing (open-to-in) conformation capable of releasing the S1-bound substrate and ions into the cytosol (lower middle structure). (B) Structural overlay of homology models of the human DAT protein (hDAT), constructed using either the LeuT (yellow ribbons) or the dDAT (light blue ribbons) crystals as structural templates. Overall, the two structures show a high degree of geometric congruence. The most profound differences are the position of the second extracellular loop region (EL2) and the orientation of TM12, which is kinked in the center of the helix in the dDAT-based structure (at P572). In the dDAT-based model, a molecule of cholesterol (sticks highlighted with a translucent blue molecular surface) is shown bound to the cavity formed by TM1, TM5 and TM7, as reported for the dDAT crystal structure. (C) Zoomed-in superposition view of both the LeuT- and dDAT-based transporter models with DA docked at the S1 site (rendered as sticks with translucent yellow and blue molecular surfaces, respectively). Residues that line the S1 binding site are labeled and rendered as thin colored sticks. (D – E) Two-dimensional molecular interaction diagrams of DA bound at the S1 site of the dDAT-based model (D) and the LeuT-based model (E). For each panel, the interaction map depicts respective DAT residues located within 4.5 Å of the bound DA molecule (hydrophobic residues are colored green and polar residues are purple). The most significant (non van der Waals) DAT/ligand interactions are indicated with dotted lines and a symbol depicting the chemistry of the interaction formed: side-chain hydrogen bond (green), main-chain hydrogen bond (blue), cation-π bond (
 +) or aromatic π-stacking (
).
+) or aromatic π-stacking (
).
As shown in Fig. 1B, the dDAT-based hDAT model (light blue) displays a general correspondence to the LeuT-based model (pale yellow), with the α-helices of the core transmembrane domains (TMs 1-11) exhibiting the highest degree of geometric congruence. The position of the substrate DA bound to the primary substrate site (S1) is also highly similar in the two models (blue vs. yellow molecular surface, respectively; see zoomed-in primary binding pocket in Fig. 1C). The DA molecule is oriented in an overall similar fashion in both models, with the amine nitrogen forming an ionic bond with D79, one of the phenyl hydroxyl groups providing hydrogen bonding with the hydroxyl of S422 in both instances, and the DA phenyl ring interacting with the aromatic ring of Y156 via π-π stacking (Figs. 1D and E). As part of a subtle difference in orientation between the two models, rather than the cation-π interaction between the charged DA amine moiety and aromatic ring of F320 observed in the LeuT-based hDAT model (Fig. 1E), an ion-dipole interaction between the DA amine and hydroxyl of S321 is seen in the dDAT model (Fig. 1D). In addition, the hydrogen bond between the second hydroxyl group of DA and S149 in the LeuT-based hDAT model (Fig. 1E) is lacking in the dDAT-based model (Fig. 1D).
The above considerations taken together indicate that the depiction of varying DAT stages shown in Fig. 1A, though based on LeuT, can be taken as an acceptable representation of overall structural changes that occur during DA transport. The uptake cycle starts with the ion/substrate-free (apo) state in which the transporter is open to the extracellular environment, the outward-facing state (Fig. 1A, top left). Na+ binding prepares the DAT for DA binding to the primary S1 site by stabilizing the transporter in the fully outward-facing state, with the extracellular gate entirely open (Na+ and Cl− bound, top right). Substrate binding to the S1 site induces closure of the extracellular gate, generating the occluded conformation (Fig. 1A, bottom right). Based on an equivocal model (Shi et al., 2008; Quick et al., 2012), a second DA molecule is depicted to bind to the S2 site in the extracellular vestibule, which may facilitate the transition of the transporter to a conformation open to the cytosol, the full inward-facing state (Fig. 1A, bottom middle). This allows dissociation of ions and substrate from the S1 site, leading to the apo inward-facing state (Fig. 1A, bottom left). In the final rate-limiting step, the DAT reverts to the outward-facing apo state allowing the initiation of another translocation cycle. Each conformation as depicted here for DAT is based on a corresponding crystal structure for LeuT (Yamashita et al., 2005; Zhou et al., 2007; Singh et al., 2007; 2008; Zhou et al., 2009; Krishnamurthy and Gouaux, 2012; Wang et al., 2013).
2.2. Atypicality and conformational status of DAT
The translocation cycle, with its transitions among different conformations of the transporter protein, offers mechanistic hypotheses for atypical properties of certain DAT ligands. One possible mechanism involves the propensity of some atypical inhibitors to induce an occluded or inward-facing conformation upon binding to the DAT. Because these more “closed” (to outward) conformations are on average markedly less probable than “open” binding conformations for which cocaine has high affinity, a compound inducing a “closed” conformation is more likely to display a slower on-rate as compared to cocaine and thus be less effective as a DAT inhibitor (see Loland et al., 2008). This hypothesis is considered again in section 3 below. A DAT inhibitor likely preferring the fully inward-facing state of DAT is ibogaine, originally reported to bind to inward-facing SERT, the serotonin transporter closely related to DAT (Jacobs et al., 2007; Bulling et al., 2012). One theory that might explain the unique activity of ibogaine is an interaction with a possible “S3 site”— located slightly below the S1 site within the cytosolic permeation pathway (Schmitt et al., 2013), with ibogaine slowly dissociating from the S3 site.
In categorizing a compound as “preferring” a more open or closed or inward conformation, it is pertinent here to consider the underlying evidence in a bit more detail. For example, cocaine and cocaine-like compounds generally display an approximate 100-fold loss in potency for the inward-facing mutant Y335A compared with wild-type DAT consonant with their preference for the outward-facing conformation prevalent in wild-type DAT (Loland et al., 2008). Conversely, benztropine or rimcazole analogs generally only show a 7- to 38-fold loss in potency for Y335A. This decrease in potency is a “preference” only in as much as cocaine-like stimulants show a substantially greater decrease in potency with the closed conformation. Alternatively it could be said that the BZT and rimcazole analogs are more tolerant to DAT conformation than are the cocaine-like stimulants (Tanda et al., 2013). As discussed by Loland et al. (2008) a reasonable interpretation is that none of these compounds stabilize a conformation identical to the most dominating conformation of Y335A, but rather stabilize conformations that are more similar to Y335A than those promoted by the cocaine analogs. Thus, the tools used only allow us to suggest that the compounds “prefer” an inward-facing conformation more-so than cocaine analogs, a subtlety that can be lost by stating they “prefer” inward DAT. In the absence of a better terminology, the current text will use the simplified statements regarding conformational “preference” of compounds.
2.3. Cholesterol and DAT conformations
Outward-facing conformations of DAT have been shown to be stabilized by transport blockers such as cocaine (as described in the prior section, and see also Chen and Justice, Jr., 1998 and Chen et al., 2000), but also by a high cholesterol content in the membrane microenvironment (Hong and Amara, 2010). Key observations from our lab as well as others have shown that DAT is associated with cholesterol-rich membrane domains, and its function is regulated by membrane cholesterol (Adkins et al., 2007; Foster et al., 2008; Cremona et al., 2011; Jones et al., 2012). The association of DAT with cholesterol was confirmed by the recent crystal structure of the Drosophila DAT (dDAT) bound with nortriptyline, a tricyclic antidepressant and transport inhibitor (Penmatsa et al., 2013). Docking of cholesterol in the hDAT model based on dDAT finds cholesterol in the same position as in the crystal: Cholesterol binds to an external hydrophobic pocket formed by transmembrane (TM) helices 1a, 5, and 7 (Fig. 1B). Molecular modeling studies comparing the structures of outward-facing dDAT with inward-facing LeuT suggest a dynamic role of cholesterol binding during the transport cycle. Because repositioning of TM1a is proposed as necessary for the transporter to transition to an inward-facing conformation, steric interference by cholesterol binding hinders the rearrangement of TM1a, thereby stabilizing the transporter in an outward-facing conformation (Penmatsa et al., 2013). This proposed mechanism of action by cholesterol is in agreement with the pharmacological and biochemical observations reported by Hong and Amara (2010). How the dynamic interaction between cholesterol and DAT modulates the conformational cycle of the DAT and influences the binding pocket of substrates or inhibitors in the core domains of the transporter will be a fascinating topic for future study.
3. Effects of Atypical DAT Inhibitors
3.1. Dopamine transporter hypothesis and atypicality
The dopamine transporter (DAT) hypothesis originated with a highly influential paper published in Science (Ritz et al., 1987). That paper hypothesized that DAT inhibitors will have cocaine-like effects differing primarily in potency. Radioligand binding techniques were used to establish the affinity of a number of drugs for the DAT and compared those relative affinities to potencies of the drugs in self-administration procedures. There was a direct relationship between these behavioral potencies and the DAT binding affinities, and very importantly the correlation among those values was greater than that for behavioral potencies and affinity for the norepinephrine or serotonin transporters. That finding has been replicated a number of times with other behavioral effects (e.g., Bergman et al., 1989; Katz et al., 2000).
Despite a substantial amount of evidence consistent with the DAT hypothesis, there are a number of compounds that bind to the DAT and inhibit dopamine uptake, but do not share cocaine-like effects in vivo. These in vivo effects are considered closely related to the abuse liability of stimulant drugs, and include locomotor stimulation, cocaine-like discriminative-stimulus effects, self-administration, and elevations in brain dopamine levels. Some of these compounds have effects that are not only different from those of cocaine, but can block the effects of cocaine. It is our opinion that the “atypical” DAT inhibitors have potential as treatments for stimulant abuse, and that they may also provide important basic information about DAT function. Among the drugs that have atypical effects are members of several different structural classes (Figure 2). Those that have been studied in the most detail are analogs of BZT (Cogentin), and several hypotheses regarding mechanisms accounting for those atypical effects have been proposed (see Tanda et al., 2009). However drugs with atypical effects that bind the DAT are not limited to analogs of BZT. We will first describe effects of BZT analogs and the various hypotheses proposed and follow that discussion with results from other drugs that have been used to assess the various hypotheses regarding atypical DAT inhibitors derived from the study of BZT analogs.
Figure 2.

Chemical structures of representative atypical DAT ligands of various types: atypical DA uptake inhibitors (top and middle rows), C1-substituted cocaine analogs (bottom left) and substrate-like partial DA releasers (bottom right). Despite their structural and mechanistic heterogeneity, atypical DAT ligands all exhibit a similar constellation of behavioral effects when compared to classical DAT inhibitors (e.g. cocaine and methylphenidate), including attenuated locomotor stimulant activity and reduced addictive liability.
3.2. Background
In an early study (Katz et al., 1999a) the stimulant-like effects of BZT analogs with substitutions on the diphenylether system were assessed. These compounds had variations in the degree to which they increased locomotor activity, though none of the compounds stimulated activity to as great an extent as cocaine (Fig. 3). In particular fluoro-substituted compounds produced stimulation that was less than that produced by cocaine, but these compounds were among the most effective of the BZT analogs. In contrast, chloro-substituted compounds, which had affinities for the DAT that were similar to those of the fluoro-analogs were generally less effective. Additionally, there were a number of other analogs with various other substitutions that were much less effective than cocaine. The BZT analogs were also tested in rats trained to discriminate cocaine from saline injections with a similar outcome: the compounds were typically less effective than cocaine and there were differences among them with regard to their effectiveness in producing partial substitution for cocaine. Further several BZT analogues were studied in rhesus monkeys trained to self-administer cocaine (e.g. Woolverton et al., 2000; 2001). When various BZT analogs were substituted for cocaine, none of the compounds maintained response rates as high as those maintained by cocaine, and in some subjects response rates were not greater than those obtained with vehicle injections. Similarly, rats trained to self-administer cocaine did not self-administer several tested BZT analogs (Hiranita et al., 2009). Further, pretreatments with BZT analogs (JHW 007, AHN 2-005) produced dose-related decreases in the maximum self-administration of cocaine, as evidenced by a shift downward in the cocaine dose-effect curve (Fig. 4). These downward shifts in the cocaine self-administration dose-effect curve differ substantially from those obtained with standard DAT inhibitor (e.g., methylphenidate, Fig. 4) that produce dose-related leftward shifts in the cocaine dose-effect curve (Hiranita et al., 2009). Similar changes in cocaine self-administration were obtained with other standard DAT inhibitors (Schenk, 2002; Barrett et al., 2004). These data suggest that the BZT analogues are acting at the DAT in a way that is different from the way in which the standard DAT inhibitors act. Similar effects of BZT treatment were obtained in subjects that were trained to self-administer methamphetamine, though not in subjects trained to self-administer heroin or the dissociative anesthetic, ketamine (Hiranita et al., 2014). These outcomes suggest that the atypical effects of these compounds on self-administration are specific to drugs with actions mediated by the DAT.
Figure 3.

Dose-dependent effects of 3α-diphenylmethoxytropane analogs on locomotor activity in mice. Ordinates, horizontal activity counts after drug administration. Abscissae, dose of drug in μmol/kg, log scale. Each point represents the average effect ± SEM determined in eight mice. The data are from the 30-min period during the first 60 min after drug administration, in which the greatest stimulant effects were obtained. Note that the fluoro-substituted compounds (A) were generally more efficacious than the other compounds, and that the bromo-substituted compounds (B) were generally the least efficacious. (Data are from Katz et al., 1999a; Hiranita et al. 2014)
Figure 4.
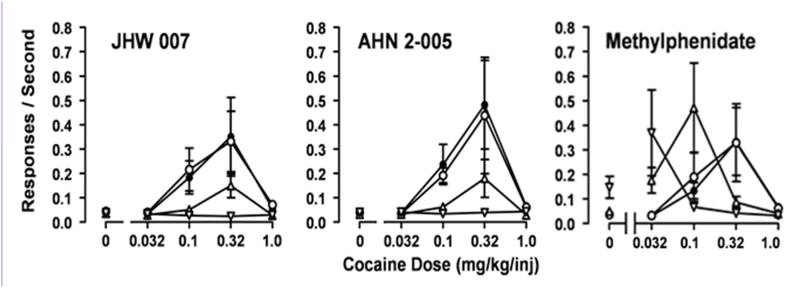
Effects of presession treatment with the N-substituted BZT analogs, JHW 007 and AHN 2-005, or methylphenidate on cocaine self-administration. Each point represents the mean ±S.E.M. (n = 6–10). JHW 007, AHN 2-005, or methylphenidate were administered orally at 300, 240 or 60 min before sessions, respectively. Filled symbols represent the intravenous self- administration of cocaine after a pre-session administration of vehicle. Open symbols represent increasing doses of the pretreatments administered by gavage. Doses of JHW 007 or AHN 2-005 were 10.0 (O), 32.0 (Δ), or 100.0 (∇) mg/kg, p.o. Doses of methylphenidate were 3.2 (O), 10.0 (Δ), or 32.0 (∇)mg/kg, p.o. Abscissae: cocaine self-administration dose in milligrams per kilogram. Each point represents the mean and standard error of the mean (n=6–11). Data are from Hiranita et al. (2014).
3.3. Pharmacological Hypotheses
There are a number of pharmacological hypotheses about the mechanisms responsible for the differences in behavioral effects between standard and atypical DAT inhibitors. As all drugs have multiple actions, the contribution of activity at off-target sites has to be considered. Other hypotheses that will be considered in more detail below are that kinetic differences between standard and atypical DAT inhibitors contribute to the differences in their effects, and that differences between the actions of standard and atypical DAT inhibitors mediated by the DA transporter contribute to differences in their effects.
3.3.1. Off-Target Effects
It is well-known that the parent compound BZT has anti-muscarinic and antihistaminic effects. These or other off-target effects might interfere with the expression of standard cocaine-like effects by the BZT analogs. Binding studies indicated that most BZT analogs are selective for the DAT among monoamine transporters, however like the parent compound many have affinity for M1 and H1 receptors (Katz et al., 2001; Zou et al., 2003; Katz et al., 2004; Hiranita et al., 2014) that may contribute to the differences between the behavioral effects of BZT analogs as compared to cocaine.
Several studies assessed the hypothesis that antagonist actions at M1 sites contributed to the atypical effects of BZT analogs (Katz et al., 2004; Tanda et al., 2009; Hiranita et al., 2014). Early studies had indicated that the non-selective anticholinergics, atropine and scopolamine, potentiated the behavioral effects of stimulant drugs (e.g., Carlton and Didamo, 1961; Scheckel and Boff, 1964) rather than the attenuation suggested by the hypothesis. The potentiation of effects of cocaine by anticholinergics was replicated with atropine and scopolamine in drug discrimination procedures (Katz et al., 1999b). Further, compounds with reduced M1 affinity retained atypical effects (Katz et al. 2004; Li et al., 2011). Similarly the more selective M1 muscarinic antagonists trihexyphenidyl and telenzepine dose-dependently and selectively potentiated the effects of cocaine on DA levels in the shell as compared to the core of the NAc and the medial pre-frontal-cortex (Tanda et al., 2007). Increases in DA in the shell of the NAc have been related the abuse liability of cocaine (Pontieri et al., 1995) and other drugs abused by humans (Tanda et al., 1997). Therefore, the potentiation of the effects of cocaine on DA levels selectively in the NAc shell is inconsistent with the hypothesis that M1 antagonist effects interfere with the reinforcing effects of BZT analogs. In general, M1 antagonist activity does not account for differences between the atypical BZT analogs and standard DAT inhibitors (Tanda et al., 2009).
As the parent compound also has histamine antagonist effects, histamine blockers have been tested alone and in combination with cocaine in rats trained to discriminate cocaine from saline injections (Campbell et al., 2005). The selective H1 antagonists, promethazine and triprolidine, did not modify the subjective effects of cocaine at any dose tested, while the H1 antagonists, chlorpheniramine and mepyramine, potentiated the subjective effects of cocaine. The effects of these latter two agents were likely mediated by their activity as DAT inhibitors (Tuomisto and Tuomisto, 1980; Bergman and Spealman, 1986; Tanda et al., 2008). In microdialysis studies, i.v. administration of diphenhydramine, and the enantiomers of chlorpheniramine, dose-dependently and selectively elevated DA levels in the shell as compared to the core of the NAc (Tanda et al., 2008). Together these results show that affinity for both H1 and DAT in diphenhydramine and chlorpheniramine does not prevent certain cocaine-like behavioral effects. As this applies to BZT analogs, it suggests that their H1 antagonist actions are probably not responsible for their reduced cocaine-like behavioral effects in animal models of drug abuse.
To further pursue the off-target action hypothesis, several BZT analogs were screened for effects at multiple mammalian receptor sites (31 or 35 sites in Katz et al., 2004 or Li et al., 2011, respectively). The initial focus was on sites at which all compounds tested uniformly had activity that was less than 10 μM, or within 100-fold of the compound's affinity at the DAT, with the 100-fold parameter chosen based not entirely arbitrarily on a typical displacement curve reaching saturation within a 100-fold range of concentrations.
3.3.1.1 Dopamine D2 Receptors
Among the sites identified were D2 dopaminergic receptors. The hypothesis that activity at D2 dopaminergic receptors could contribute to atypical DAT effects of compounds was examined by assessing the antagonism of cocaine-induced locomotor stimulation by JHW 007 in D2 DAR knockout and wild-type mice, rather than with novel compounds. Although the locomotor-stimulant effects of cocaine were diminished in DA D2R knockout mice compared to that in their WT littermate controls, pretreatment with JHW 007 blocked the stimulant effects of cocaine equally in both knockout and wild-type mice (Desai et al., 2014). Therefore the data suggest that a D2 dopamine receptor is not necessary for the atypical DAT inhibitor effects of JHW 007.
3.3.1.2 Allosteric Modulation of CB1 Receptors
Previous studies suggested that the isoxazol phenyltropane derivative, RTI-371 [3β-(4-methylphenyl)-2β- [3-(4-chlorophenyl)isoxazol-5-yl]tropane] (Fig. 2) has effects similar to other atypical DAT-inhibitors. For example, RTI-371 failed to stimulate locomotor activity in mice across a range of behaviorally active doses and did not substitute in rats trained to discriminate cocaine from saline injections (Carroll et al., 2004). Further, a preliminary report indicated that RTI-371 blocked cocaine-induced locomotor stimulation (Navarro et al., 2009). Finally, because both RTI-371 and JHW007 produced positive-allosteric modulation of CB1Rs, and CB1Rs have been implicated in the neurobiology of cocaine addiction (see Tanda, 2007 for review), Navarro et al. (2009) suggested that the atypical effects of both of these compounds were a result of a positive allosteric modulation of CB1Rs, as indicated by the increased efficacy of the CB1 agonist, CP 55940 in a calcium mobilization assay. From those results Navarro et al. suggested that enhanced endocannabinoid neurotransmission may contribute to the cocaine-antagonist effects observed with these atypical DAT inhibitors. This notion was tested with CB1R wild-type and knockout mice. Cocaine stimulated locomotor activity in both wild type and knockout mice. Additionally, co-injection of JHW 007 significantly blunted the locomotor stimulant effects of cocaine in both CB1R wild-type and knockout mice (Desai et al., 2014). Thus it appears that positive allosteric modulation of CB1Rs is not necessary for the atypical effects of either of these compounds.
3.3.1.3 Sigma Receptors
A number of previous studies indicated that σ-receptor antagonists can block several of the effects of cocaine, including locomotor stimulation, sensitization, and place conditioning (see reviews by Matsumoto, 2009 and Katz et al., 2011), though not its self-administration (Martin-Fardon e et al., 2007; Hiranita et al., 2011). However, the carbazole derivative, rimcazole which is a sigma receptor antagonist, blocked cocaine self-administration (Hiranita et al., 2011), as did two of its analogs, SH 3-21 and SH 3-28 (Figure 2). Rimcazole is unique as a σR antagonist as it has affinity for the σR as well as the DAT (Izenwasser et al., 1993; Valchar and Hanbauer, 1993). Therefore, it was noteworthy that BZT analogs also have affinity for σRs (Katz et al., 2004; Li et al., 2011).
To assess whether these mechanisms could produce effects similar to those of atypical DAT inhibitors, combinations of several selective σR antagonists and DAT inhibitors were administered to rats trained to self-administer cocaine, and effects like those of rimcazole were obtained (Hiranita et al., 2011). That effects similar to those of rimcazole were obtained with several different combinations of σR antagonists and DAT inhibitors indicates that the decreases in self-administration were not isolated idiosyncratic effects. Additionally, the decreases in cocaine self-administration were obtained at dose combinations that did not decrease comparable responding maintained by food reinforcement, a selectivity reminiscent of that seen with the atypical DAT inhibitors. Combinations of σR antagonists and DAT inhibitors were also effective against methamphetamine self-administration (Hiranita et al., 2014).
Although many of the BZT analogs, as well as the carbazole analogs of rimcazole tested to date, have sigma receptor antagonist effects, the isoxazol phenyltropanes, RTI-336 and RTI-371, had relatively low affinity at sigma receptors (Hiranita et al., 2014). As discussed above, several characterizations of the in vivo effects of RTI-371 have indicated atypical DAT inhibitor effects (e.g., Carroll et al., 2004; Navarro et al., 2009). RTI-336 is a structural isomer of RTI-371 and has been the more fully studied of the two. There are several reports that pretreatment with RTI-336 decreases self-administration of cocaine doses that maintained maximal or near maximal rates of responding (e.g., Haile et al., 2005; Howell et al., 2007). However, there are also reports that RTI-336, in contrast to RTI-371, fully substitutes in rats trained to discriminate cocaine from saline injections (e.g., Carroll et al., 2004), an effect obtained neither with RTI-371 nor most of the BZT analogs (Katz et al., 1999a).
In self-administration studies with rats (Hiranita et al., 2014), RTI-336 was self-administered at levels greater than vehicle, an effect similar to that obtained with standard DAT inhibitors. As had been reported previously (e.g., Howell et al., 2007), the rates of responding maintained were less than those maintained by cocaine, an effect likely attributable to its long duration of action relative to that of cocaine. In contrast to those effects, its structural isomer, RTI-371, was not self-administered at rates above those obtained with vehicle, an effect similar to that obtained with atypical DAT inhibitors. Most importantly, RTI-336 produced dose-related leftward shifts in the cocaine self-administration dose effect curve, whereas RTI-371 produced dose-related decreases in the maximum self-administration of cocaine, an effect reminiscent of that produced by the BZT and rimcazole analogs. Confounding attempts to find a single mechanistic explanation for atypical DAT inhibitor activity, neither isoxazole phenyltropane had appreciable affinity for sigma receptors, Further, despite differences in their behavioral profile the structural isomers were similar to each other in their high affinities for the DAT and lower affinities for sigma receptors (Hiranita et al., 2014).
Interestingly, the most marked difference observed between the two isomers was found using in vivo binding techniques (Hiranita et al., 2014). At the highest dose of RTI-371 studied (56 mg/kg), displacement of the DAT radiolabel, [125I]RTI-121, occurred at a rate of 0.320%/min, which was approximately one-half of the rate with RTI-336 and one-fourth of the rate with cocaine. Thus these data suggest that rate of association may play a critical role in determining the difference in the effects of these two compounds. An initial hypothesis derived from studies of the analogs of BZT focused on their relatively slow rate of association compared to that of cocaine. Recently, Kohut et al. (2014) suggested that slower DAT association may create conditions favorable to desensitization of postsynaptic DA receptors that, in turn, would be less effective in transmitting DA signals. Such a desensitization process agrees with the low efficacy of atypical DAT inhibitors with regard to many of the often observed stimulant effects.
3.4. Kinetic Variables
One of the initial hypotheses about the atypical effects of BZT analogs was that the effects might be due to pharmacokinetic differences between the BZT analogs and standard DAT inhibitors. The onset of the effects of cocaine is generally much faster than that for most BZT analogs (Tanda et al., 2005). In vivo binding studies confirmed the slow rate of onset of action for JHW007 (Desai et al., 2005). Coupled with pharmacokinetic studies indicating that many of the BZT analogs appear in brain within minutes of injection (Raje et al., 2003) at concentrations exceeding their in vitro Ki values, the studies suggest a slow association rate for BZT analogs that was confirmed in vitro (Kopajtic et al., 2010). One hypothesis is that a slow association rate allows for compensatory mechanisms that blunt the effects of cocaine (Kohut et al. 2014). A DAT inhibitor with a faster association with the DAT would not allow for that compensatory change.
A recent study examined a group of BZT analogs that have a relatively fast onset of behavioral effects (Li et al., 2011). All of the compounds studied bind to the DAT and inhibit dopamine uptake in vitro, and had maximal behavioral effects within 30 minutes of injection. However, those effects were generally not characteristic of standard DAT inhibitors. All but one of these compounds (JHW 013) produced only dose-related decreases in locomotor activity, and JHW013 had stimulant effects that were substantially reduced compared to those of cocaine. Thus, at the present time kinetic differences between BZT analogs and standard DAT inhibitors do not appear to fully account for their atypical effects.
3.5. DAT Structure and Function
Several studies addressed whether structural and functional features of the DAT can be related to the atypical actions of BZT analogs. The first of these studies(Reith et al., 2001) reported that cocaine-like drugs produce a DAT conformation that differs from that produced by BZT analogs. Several other studies followed (e.g., Chen et al., 2004; Ukairo, Bondi et al., 2005) showing various point mutations in the DAT changed affinity of standard DAT inhibitors in a way different from the way in which affinity was changed for atypical DAT inhibitors, suggesting different binding domains for these compounds. Loland et al. (2008) found that those compounds that had DAT affinity and promoted a conformation of the DAT open to the extracellular space were like cocaine in vivo, both in a cocaine discrimination procedure as well as in stimulating locomotor activity. In contrast, those compounds that promoted a conformation of the DAT open to the cytosol did not fully substitute in subjects discriminating cocaine, and were less effective than cocaine in stimulating locomotor activity.
In a recent study (Reith et al., 2012), several C-1 substituted analogs of cocaine (see Figure 2) were studied in vitro and in vivo. Despite CNS penetration, none of the compounds stimulated locomotor activity in mice suggesting atypical DAT inhibitor effects. Despite these behavioral differences from cocaine, in silico docking of two of the compounds exhibited, like cocaine, a preferential interaction with an outward facing DAT conformation. Thus these compounds showed differences from cocaine with regard to their in vivo stimulatory effects but were unlike the BZT analogs with regard to their predicted interaction with the DAT. Interestingly, several of these cocaine analogs had some affinity for sigma receptors, suggesting that their atypical effects may originate from activity similar to that of rimcazole (see above). However, if their sigma activity is like that of cocaine, they will be agonists rather than antagonists, whereas rimcazole is a sigma receptor antagonist. Nonetheless, these compounds had effects that differed substantially from cocaine for which accounts must be made in any unified hypothesis regarding mechanism of atypical DAT inhibitors.
Results with several DAT inhibitors on hDAT conformation shown in Figure 5A confirm and extend previous findings. In the present studies hDAT was point mutated, replacing threonine-316 with cysteine and replacing cysteine-306 with alanine, yielding a single cysteine residue in the extracellular vestibule when DAT was in an outward-facing conformation. Reactivity to maleimide-PEO2-biotin was used as a reporter of DAT conformational status as previously validated (Hong and Amara, 2010). Uptake of dopamine in cells transfected with this mutant DAT was similar to that in cells transfected with wild-type DAT, indicating that the mutation did not interfere with functional DA transport. Incubation with cocaine (100 μM) and methylphenidate increased the cysteine-316 accessibility, suggesting that these compounds induce an outward-facing conformation of DAT. In contrast, BZT, GBR12909 and the stereo isomers of modafinil decreased cysteine accessibility, whereas rimcazole was without substantial effects.
Figure 5.

Different effects on the cysteine accessibility of DAT T316C/C306A induced by various inhibitors. (A) Hong et al., unpublished results. (B) Adapted from Hiranita et al, 2014. HEK293 cells stably transfected with T316C/C306A human DAT were labeled with maleimide-PEO2-biotin in the presence of DAT inhibitors. Biotinylated DAT proteins were enriched with avidin beads from cell lysates and detected with DAT-specific antibodies. Mean densities of DAT bands were quantified using the NIH ImageJ software and normalized to percent of vehicle (error bar is SEM). Representative blot from n = 3 - 6 experiments. Quantified DAT band densities were analyzed by one way ANOVA with post hoc Dunnett's test or Fisher's least significant difference test. **, P<0.01, *P<0.05 compared with vehicle. See method details in Hong and Amara (2010) and (Hiranita et al., 2014).
As this mutant DAT replicates the earlier findings on DAT ligands and conformation, it was employed to examine the generalizability of the relation between atypical effects and DAT conformational status using the isoxazol derivatives of cocaine (Hiranita et al., 2014). Incubation with cocaine (100 μM), RTI-336 (1.0 μM), and RTI-371 (1.0 μM) significantly increased DAT labeling (Fig. 5B), indicating that the side chain accessibility of residue 316 in the middle of TM6a was altered in a similar way by the binding of these drugs to DAT. In contrast, incubation with BZT (100 μM) and its analog JHW007 (1.0 μM) reduced DAT labeling. These results indicated that both RTI-371 and its structural isomer RTI 336 induced conformational changes in the DAT like those produced by cocaine. Those results suggest that conformational status of the DAT does not uniformly confer atypical DAT inhibitor effects on drugs inducing those conformations.
Several previous studies indicated that the DAT inhibitor, modafinil and its enantiomers, induces an inward-facing conformation of the DAT (Schmitt and Reith, 2011; Loland et al., 2012). Those effects were replicated using the DAT T316C/C306A mutant (Fig. 5A). As outlined above, that outcome is thought to be predictive of atypical effects, and indeed modafinil is reported to have low abuse liability (Minzenberg and Carter, 2008) and potential efficacy as a treatment for cocaine abuse (Hart et al., 2008; Dackis et al., O'Brien, 2012). Other studies directed at treatment of stimulant abusers have reported mixed results (see review by Mereu et al., 2013), although it is clear that modafinil does not produce a cocaine-like ‘high’ in humans and has little abuse liability of its own, even in frequent cocaine users (Vosburg et al., 2010). However, in contrast to the effects of other compounds promoting an inward-facing DAT conformation, modafinil fully substituted for cocaine in mice trained to discriminate cocaine (10 mg/kg) from saline injections (Loland et al. 2012), stimulated locomotor activity, though to a lesser extent than cocaine (Cao et al., 2010), and increased DA concentrations in the nucleus accumbens shell, though also to a lesser extent than cocaine and with an apparent plateau across the doses below those producing acute toxicity (Loland et al., 2012). There also have been mixed results in studies of self-administration using laboratory animals (e.g. Gold and Balster, 1996; Newman et al., 2010).
Taken together, studies of modafinil, RTI-371, and C-1 cocaine analogs have not supported the notion that DAT conformational status necessarily predicts atypical DAT effects behaviorally or clinically. Further, what constitutes atypical effects of DAT inhibitors must be nuanced as different drugs acting at the DAT can have a different spectrum of atypical effects. Perhaps the results obtained so far can be summarized by stating that there are different paths to atypicality, but that no path by itself guarantees behavioral/clinical atypicality. An elaboration of this with more detail is provided in section 5.
4. Atypical Releasers
The classical DA releasing agents amphetamine and methamphetamine are two of the few medications to show promise in controlled clinical trials as treatments for stimulant addiction(Grabowski et al., 2004; Mooney et al., 2009; Karila and Reynaud, 2009). They also decrease cocaine self-administration behavior in non-human primate models of addiction (Wojnicki et al., 1999; Negus and Mello, 2003; Negus et al., 2007; 2009). These studies provide support for using classical DA substrates (i.e., DA releasers) as potential medications for the treatment of stimulant-use disorders. Recent evidence suggests that just as DA uptake inhibitors can vary in their interactions with the DAT, DA releasers also have various effects on the DAT. If one considers amphetamine as the prototypical DA releaser, then a variety of factors can contribute to defining “atypicality” with respect to DAT substrates. These factors include: 1) reduced efficacy for releasing DA (i.e., partial release), 2) releasing activity at non-DAT transporters, especially the 5-HT transporter (SERT), 3) reduced activity at the vesicular monoamine transporter 2 (VMAT2), and 4) mixed uptake inhibition/release properties at different biogenic amine transporters (i.e., so-called “hybrid” activity). One might consider DA releasers as “atypical” if they display one or more of the above-mentioned effects, analogous to the atypical DA inhibitors discussed previously. Various combinations of these effects in single lead molecules result in a large number of theoretical DA releaser subtypes that differ from the classic DA releasing stimulants, each with their own unique therapeutic potential.
4.1 Releaser Transportability
Understanding the concept of atypical releaser activity requires an understanding of the transporter conformation cycle and the transporter-mediated release process itself, especially the fact that releaser molecules must be translocated from the extracellular space into the cytoplasm (i.e., releasers are transportable). Thus, DA releasers are DAT ligands that bind at the site of translocation and are transported along with sodium ions in the same manner as the endogenous substrate DA (see Figure 1A) (Sulzer et al., 2005; Leviel, 2011; Gowrishankar et al., 2014). Converging lines of evidence have solidified the notion that DA releasers are substrates of the transporter and once translocated, they reverse the normal direction of transporter flux to evoke release of endogenous neurotransmitters. The nature of this reversal is not well understood, but the entire process is primarily transporter-dependent and requires elevated intracellular sodium concentrations, phosphorylation of DAT, and possible involvement of transporter oligomers (Khoshbouei et al., 2003, 2004; Sitte and Freissmuth, 2010). Our investigations of the transporter activity for a large library of small molecular weight amine-containing compounds support the hypothesis that releasers are transporter substrates. Compounds were assessed in both release and uptake inhibition assays, according to protocols developed by Rothman and colleagues (Rothman et al., 2001, 2002). These protocols, carried out in synaptosomes from rat brain, take advantage of differences in how DAT ligands behave in release and uptake inhibition assays. Synaptosomes can be viewed as isolated nerve endings with DAT proteins oriented in the same manner as in intact neurons. In DAT release assays, radiolabeled transmitter (e.g., [3H]DA) is preloaded into synaptosomes prior to addition of the test drug; in DAT uptake inhibition assays, [3H]DA and test drugs are added to synaptosomes simultaneously, thereby competing for the same transporter sites. If a test compound is a DA releaser, it causes accumulated [3H]DA in either assay to move out of synaptosomes, and would display activity in both assays. By contrast, DAT uptake inhibitors cannot enter synaptosomes and induce DA release, so they would only be active in the DAT uptake inhibition assay. Therefore, test compounds which are examined in these two assay systems can be inactive in both assays, active in both assays (i.e., a releaser), or active only in the uptake inhibition assay (i.e., an uptake inhibitor).
A library of approximately 1400 phenethylamine compounds (PAL compounds) has been screened using these protocols. Among the active compounds, the smaller DAT ligands were found to be DA releasers while the sterically larger compounds were DAT uptake inhibitors. Releaser activity was further characterized for many of these structures by obtaining values for potency (i.e., EC50 values) and efficacy (i.e., % maximal release), and by running substrate reversal experiments. The latter experiments involve the inclusion of known DAT uptake inhibitors in the release assay, to verify that releasing effects of test drugs are transporter-dependent. From a medicinal chemistry perspective, our findings confirm that small compounds, similar in size to amphetamine or DA, can induce release, precisely what one would expect with a mechanism that relies on a compound being a transporter substrate.
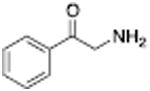
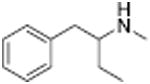
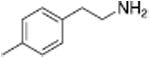
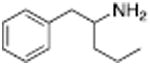
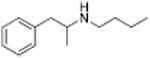
Table 1 shows the transporter activity of two sets of compounds, one a set of phenethylamine (i.e., amphetamine) analogs and the other a set of β-keto phenethylamine (i.e.,cathinone) analogs. Each set of compounds demonstrates predicable structural trends. Amphetamine is a DA releaser with an EC50 value of 8.7 nM. Modifications that increase the size of amphetamine gradually decreased release potency until the increases caused the activity to change to transport inhibition. Specifically, N-alkylation of amphetamine, going from no alkyl group (amphetamine) to methyl (methamphetamine) to ethyl (PAL-99) decreased EC50 values for release from 8.7 to 24.5 to 88.5 nM. Adding an additional methylene to form the N-propyl analog (PAL-424) caused the compound to become a DAT uptake inhibitor with an IC50 value of 1013 nM. Increasing the size even further to butyl (PAL-90) rendered the compound inactive at the DAT. Increases in the size of the 4-phenyl substituent on the phenyl ring produced a similar trend. Phenyl ring substitution of phenethylamine, from no substitution (phenethylamine) to a 4 –methyl substituent (PAL-503) to the 4-ethyl substituent (PAL-505) increased EC50 values (reduced potency) for release from 39.5 to 271 to 2087 nM. A 4-isopropyl substituent (PAL-595) rendered the compound inactive. An increase in the size of the α-alkyl group produced the same trend. PAL-426, with an α-ethyl substituent, had an EC50 value of 225 nM whereas PAL-550, with an α-propyl substituent, changed activity to predominantly uptake inhibition, with an IC50 value of 2596 nM (Table 1).
Table 1. Dopamine (DA) transporter uptake inhibition and substrate releasing effects of a series of phenethylamines as determined in rat brain synaptosomes.
| Name | Structure | DA UI IC50 (nM) | DA Release EC50 (nM) | Name | Structure | DA UI IC50 (nM) | DA Release EC50 (nM) |
|---|---|---|---|---|---|---|---|
| Phenethylamine |
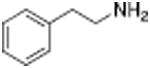
|
39.5 ± 1.8 | PAL-27 |
|
208 ± 13 | ||
| Amphetamine |
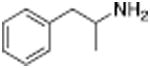
|
8.7 ± 1.2 | Cathinone |
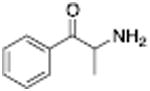
|
83.1 ± 9 | ||
| Methamphetamine |
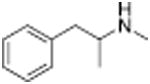
|
24.5 ± 2.1 | Methcathinone |
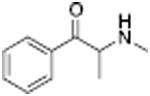
|
49.9 ± 3.1 | ||
| PAL-99 |

|
88.5 ± 9.6 | PAL-434 |
|
46.8 ± 4.0 | ||
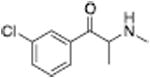
| PAL-426 |
|
225 ± 2.1 | Mephedrone |
|
49.1 ± 3.1 | ||
| PAL-503 |
|
271 ± 17 | Methylone |
|
133.0 ± 11 | ||
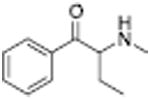
| PAL-505 |
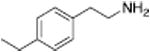
|
2087 ± 195 | PAL-429 |
|
411 ± 98 | ||
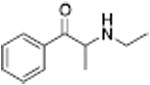
| PAL-424 |
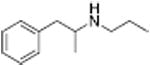
|
1013 ± 101 | N-Ethylpropion |
|
1014 ± 80a | ||
| PAL-550 |
|
2596 ± 408 | PAL-359 |
|
IA | ||
| PAL-90 |
|
IA | MDPV |
|
4.1 ± 0.06 | ||
| PAL-595 |
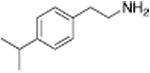
|
IA | Bupropion |
|
658 ± 178 |
Release EC50 and Uptake Inhibition IC50 values are reported as means ± SD and are the result of n= 3 performed in triplicate.
UI = Uptake Inhibition; IA = inactive with IC50 >10,000 nM. Cells are left blank where the major property of the compound is in the adjacent cell (releasing activity in top portion of table, or uptake inhibitory activity in the bottom portion).
From Yu et al., 2000.
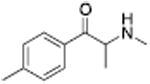
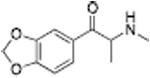
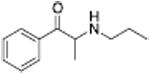
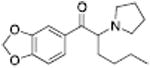
Analogous trends in structure-activity relationships were observed in a series of cathinone analogs (Table 1). Methcathinone is a DAT substrate and DA releaser with an EC50 of 49.9 nM. Its ethyl analog, α-ethylmethcathinone (PAL-429), is an order of magnitude weaker as a DAT releaser, with an EC50 value of 411 nM. Substitution with an N-ethyl group (N-ethylpropion) changed the compound to an uptake inhibitor with an IC50 value of 1014 nM. N-propylation (PAL-359) rendered the compound in active. Phenyl ring substitution does not seem to affect potency as much in the cathinone series as it does in the phenethylamine series. The 3-chloro (PAL-434) and 4-methyl (mephedrone) substituted methcathinones were approximately the same potency as methcathinone, with EC50 values of 46.8 and 49.1 nM respectively. However, the slightly larger 3,4-methylenedioxy analog (methylone) had a reduced releaser potency with an EC50 value of 133.0 nM. Both MDPV and bupropion, which feature larger moieties in 2 or 3 locations around the cathinone core are uptake inhibitors with IC50 values of 4.1 nM and 658 nM respectively. It is interesting to note that the cathinone series, bearing the β-ketone functional group, is also weaker compared to the analogous compound in the phenethylamine series, further demonstrating the size trend: Cathinone is a weaker releaser than amphetamine; methcathinone is weaker than methamphetamine; PAL-429 is weaker than PAL-426; and PAL-359 is inactive while PAL-424 retains DAT ligand activity.
In each series as the scaffold increases in size, there is a structural cutoff point where the releaser activity converts to being an uptake inhibitor. Presumably at that point, the compound still binds to the DAT at the site of translocation (S1), but is too large to undergo translocation and thus becomes an uptake inhibitor. In addition, the releasing potency weakens as the size of the structure increases and moves towards the “edge” of the pharmacophore, even before the compound becomes an uptake inhibitor. This observation suggests that certain structural features could alter the kinetics of the rate-limiting step of the release process. Similar trends are observed in our data (unpublished) with both the SERT and the norepinephrine transporter (NET). The implication from these observations is that the rate-limiting step of transporter-mediated release might involve binding, translocation, or a combination of the two, since those events must occur before reversal of the transporter to induce transmitter release. If true, these release structure-activity data contain substantial information relevant to the mechanism of substrate translocation. Conceptually, the trends in Table 1 suggest a generic pharmacophore as shown in Fig. 6, in which the theoretical space for a releaser is in the center, and structural expansion in any direction eventually converts the compound to an uptake inhibitor. Detailed molecular modeling of substrate activity at the DAT and SERT has been carried out using phenylpiperazine and phenethylamine analogs which are releasing substrates (Severinsen et al., 2012; Seddik et al., 2013). Other models are currently being developed.
Figure 6.

Generic pharmacophore for biogenic amine transporter ligands. Note that transportable substrate ligands exhibit size constraints defined by the red circle. Functional groups attached to the nitrogen, α-carbon or phenyl ring that extend beyond the “edge” of the pharmacophore will generate partial substrates, transporter blockers or be inactive.
4.2 Hybrid Transporter Profiles
The concept that releasers depend on transportability can explain differences in selectivity of releasers among the biogenic amine transporters and lead to unique transporter ligand profiles. For example, the structural features needed for transport by the DAT differ from those required for transport by the SERT, so schematically, the release pharmacophores for both transporters overlap but are offset from each other and differ in shape. On their edges, the pharmacophores diverge in activity. These differences enable the discovery of compounds that are substrate releasers at one transporter while being uptake inhibitors at a different transporter. Chemical structures with mixed uptake inhibitor/releaser activity have been termed “hybrids”, examples of which are shown in Table 2 (Blough et al., 2014). N-Ethylpropion was the first hybrid compound identified, with a DAT uptake inhibition IC50 value of 1014 nM and a 5-HT release EC50 value of 2118 nM (Yu et al., 2000). Structurally, N-ethylpropion lies right past the point where a scaffold series changes from being a DAT substrate to a DAT inhibitor (non-substrate) but is still a substrate of the SERT leading to release (see Table 1, right-hand side). A series of related N-cyclopropylcathinone analogs was found to also have similar hybrid activity (Table 2), including the 3-chloro (PAL-433), the 3-bromo (PAL-586), the 3,4-dichloro (PAL-787) and the 3-chloro-4-methyl (PAL-820) analogs. All retain the beta-ketone and N-cyclopropyl groups but vary in phenyl ring substitution. These compounds became the first class of hybrid transporter ligands to be explored in detail (Blough et al., 2014). Nearly all combinations of DAT, SERT, and NET hybrids have been observed.
Table 2. Comparison of dopamine transporter (DAT) uptake inhibition and serotonin (5-HT) releasing effects for a series of N-alkylpropiophenones (cathinones) as determined in rat brain synaptosomes.
| Name | Structure | DAT UI IC50 (nM) | 5-HT Release EC50 (nM) |
|---|---|---|---|
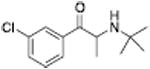
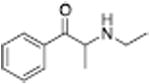
| N-Ethylpropion |
|
1014 ± 80a | 2118 ± 98a |
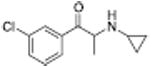
| PAL-433 |
|
533 ± 61 | 1328 ± 148 |
| PAL-586 |
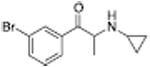
|
386 ± 29 | 621 ± 141 |
| PAL-787 |
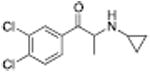
|
425 ± 22 | 356 ± 96 |
| PAL-820 |
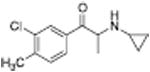
|
592 ± 31 | 181 ± 31 |
Release EC50 and Uptake Inhibition IC50 values are reported as means ± SD and are the result of n= 3 performed in triplicate.
DAT/SERT hybrids are important because mixed action DAT/SERT ligands may differ in therapeutic potential when compared to dual DA/5-HT releasers or dual DA/5-HT uptake inhibitors. In all cases, acute drug effects would involve elevation of extracellular DA and 5-HT in the brain, but the extent of transmitter increase could differ considerably. In vivo microdialysis studies have shown that transporter inhibitors induce much smaller increases in extracellular neurotransmitter when compared to effects of transporter releasers (e.g., see Gundlah et al., 1997) and Rothman and Baumann, 2002). The neurochemical effects of transporter inhibitors are dependent upon ongoing cell firing (i.e., effects are impulse-dependent) and exocytotic transmitter release (i.e., effects are calcium-dependent), whereas the effects of transporter releasers are not. As such, neurochemical effects of transport inhibitors are sensitive to autoreceptor-mediated negative feedback effects which serve to limit the magnitude of drug-induced transmitter increases. The rate of increase in extracellular neurotransmitter evoked by releasers is likely faster than that of inhibitors because it is a direct effect, as opposed to the indirect blocking of neurotransmitter uptake. Because releasers are transported into cells along with sodium ions, they induce inward current and facilitate channel-like transporter activity; by contrast, uptake inhibitors block transporter-mediated currents (Sonders et al., 1997; Sitte et al., 1998). When administered chronically, DA releasers and DA uptake inhibitors have dissimilar effects on trafficking of DAT and downstream signaling, in some cases the effects being completely opposite (Zahniser and Sorkin, 2004). Uptake inhibitors like cocaine upregulate surface DAT expression while DA releasers have the exact opposite effect (Daws et al., 2002; Cervinski et al., 2005 and see review by Schmitt et al., 2013). These differences in the acute and chronic effects of releasers versus blockers may lead to differences in therapeutic potential, as exemplified by NET uptake inhibitors which are effective antidepressants whereas NE releasers are not (Wong et al., 2000).
These differences suggest the need to explore both release and uptake inhibition when analyzing the transporter activity of small phenyl amines as drug candidates. PAL-433 was originally synthesized as part of a series of bupropion analogs (Carroll et al., 2009). It was originally found to be a DAT/SERT uptake inhibitor, but this study did not include neurotransmitter release assays so the possibility that the PAL-433 could have DA or 5-HT release activity was not explored. When studied using the Rothman protocol, PAL-433 was found to be a DAT uptake inhibitor with an IC50 value of 533 nM and was a 5-HT releaser with an EC50 of 1328 nM. Thus, the compound was initially mischaracterized. This finding highlights the importance of including both release and uptake inhibition assays when determining the profile of transporter activity for ligands with low molecular weight.
4.3 Partial Releasers
Another phenomenon observed in the study of neurotransmitter releasers is low-efficacy “partial” releasing effects of certain ligands (Rothman et al., 2012). Partial release can be viewed as analogous to partial agonism at G protein-coupled receptors. Partial releasers are transporter substrates that do not release the maximum releasable pool of neurotransmitter even at the highest concentrations, examples of which are shown in Table 3. A DA partial releaser like MDEA has an EC50 value for release of 622 nM, but with an EMAX of only 61%. Its N-methyl analog MDMA or the analog without the 3,4-methylenedioxy group (PAL-99) are both more potent releasing substrates with EC50 values of 108 and 115 nM, respectively, and are full releasers with EMAX values of 103% and 111%. A similar trend was observed for a series of compounds based on PAL-287, the initial proof-of-concept compound in this project. PAL-287 is a potent DA releaser with an EC50 value of 12.6 nM and an EMAX value of 102%. The N-methyl analog PAL-1046 has an EC50 value of 10 nM and an EMAX value of 101%. The N-ethyl analog, PAL-1045, is slightly less potent, but with an EMAX value of only 78%. The phenomenon of partial release is not just observed with phenethylamines. 2-Methylphenylpiperazine (PAL-169) is a DA releaser with an EC50 value of 296 nM and an EMAX value of 90%. 2,3-Dimethylphenylpiperazine (PAL-218) is a weaker DA substrate with an EC50 of 1207 and releases only 66% of the releasable pool of DA. Partial release can also be observed at SERT and NET (Rothman et al., 2010).
Table 3. Dopamine (DA) uptake inhibition and releasing effects for a series of phenethylamines that display the transition from full efficacy, to partial efficacy, to uptake inhibition as determined in rat brain synaptosomes.
| DAT UI | DA Release | |||
|---|---|---|---|---|
| Name | Structure | IC50 (nM) | EC50 (nM) | EMAX |
| PAL-287 |
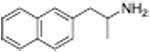
|
12.6 ± 1 | 102 ± 1 | |
| PAL-1046 |
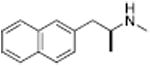
|
10 ± 1 | 101 ± 3 | |
| MDMA |
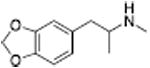
|
108 ± 10 | 103 ± 2 | |
| PAL-99 |
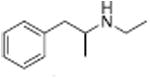
|
115 ± 35 | 111 ± 13 | |
| PAL-169 |
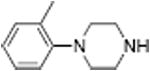
|
296 ± 20 | 90 ± 1 | |
|
| ||||
| PAL-1045 |
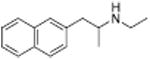
|
46 ± 11 | 78 ± 5 | |
| MDEA (PAL-193) |
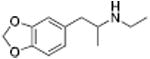
|
622 ± 121 | 61 ± 2 | |
| PAL-218 |
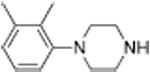
|
1207 ± 118 | 66 ± 1 | |
|
| ||||
| PAL-424 |
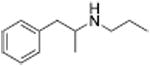
|
1013 ± 101 | ||
| MDPV |
|
4.1 ± 0.06 | ||
| PAL-233 |
|
9523 ± 436 | ||
Release EC50 and Uptake Inhibition IC50 values are reported as means ± SD and are the result of n= 3 performed in triplicate. Otherwise details are as for Table 1.
From a therapeutic point of view, partial releasers share neurochemical similarities with uptake inhibitors since the levels of extracellular neurotransmitter evoked by these drugs could be much lower than a classical full efficacy releaser. Further, the rate of neurotransmitter elevation with a partial releaser might be kinetically slow like an uptake inhibitor, but such compounds are nonetheless transporter substrates and will affect biological systems like a releaser after chronic administration. Partial release may be related to a substantial decrease in the rate of translocation of drug molecules through the transporter, but this hypothesis remains to be tested. Another possibility is that partial releasers such as N-ethyl-amphetamines and similarly sized substrate-like molecules briefly stall the DAT conformational cycle by interacting with a possible “S3 site” — located slightly below the S1 site within the cytosolic permeation pathway (see section 2.3). That is, the increased steric bulk of partial substrate-type DAT ligands causes them to dissociate from the cytosolic permeation pathway more slowly than smaller full substrates, resulting in a slower rate of both DA uptake and release. All of the partial releasers identified thus far are structurally at the edge of the release pharmacophore. When compared to similar structures, they all lie between the size of a fully efficacious releaser and a pure uptake inhibitor as shown in Table 3. Sterically larger compounds such as PAL-424 and MDPV are both uptake inhibitors. 4-Methylphenylpiperazine (PAL-233) is also a DAT inhibitor with an IC50 of 9523 nM. So, these partial releasers appear to be small enough to be substrates, but just barely get transported. Partial releasers may have substantially different therapeutic potential when compared to full efficacy releasers. In vivo microdialysis studies in rats have shown that extracellular DA concentrations increase after systemic administration of the DA partial releaser PAL-1045 as shown in Fig. 7, but as predicted, the magnitude of effect on dialysate DA is lower than that of a full efficacy releaser PAL-1046. Effects of the partial releaser on locomotor stimulation were also smaller when compared to the full efficacy releaser (Fig. 7). Partial releaser pharmacology greatly increases the potential for mechanistic combinations among transporter ligands. For instance, one can envision a DA full releaser/5-HT partial releaser combination or a DA partial releaser/5-HT uptake inhibitor hybrid. As noted above, each combination could have its own unique therapeutic potential.
Figure 7.

Effects of DAT releasers on extracellular dopamine (DA) and horizontal locomotor activity (HLA) in male rats. Rats undergoing microdialysis in the n. accumbens received sequential intravenous doses of the fully efficacious releaser PAL 1046, the partial releaser PAL-1045 or saline vehicle. Arrows indicate time of injections (1 or 3 mg/kg i.v.). Dialysate DA concentrations were assayed by HPLC-ECD. Data are mean ± SEM for n=6-7 rats/group.
4.4 The Effects of 5-HT Release
The in vivo pharmacology of DA releasers can be complicated by effects of concurrent 5-HT and/or NE release. One of the first observations made when exploring potential releaser scaffolds was that phenotypic behavioral effects of DA releasers, like locomotor stimulation, seemed to decrease as 5-HT release potency increased. Our initial proof of concept compound, PAL-287, was a potent non-selective releaser with DA, 5-HT, and NE releasing EC50 values of 12.6 nM, 3.4 nM, and 11.1 nM, respectively (Rothman et al., 2005). Interestingly, the compound was a weak locomotor stimulant and lacked strong reinforcing effects, when compared to a selective DA/NE releaser such as amphetamine (200-300 fold selective for DA/NE over 5-HT release). These observations were similar to the preclinical effects of the combined administration of fenfluramine and phentermine, in which the 5-HT releaser fenfluramine reduces stimulant effects of the DA releaser phentermine (Rothman et al., 1998; Rea et al., 1998; Baumann et al., 2000). In vivo microdialysis experiments in rats showed that PAL-287 generally induced elevations of monoamines concurrent with its in vitro substrate activity, but released much less DA than expected (Rothman et al., 2005). This led to speculation that 5-HT release activity might be reducing DA release. A series of four ring-substituted amphetamine analogs was developed which have similar DA releasing potencies with EC50 values ranging between 24.2 nM and 51.5 nM, but which vary greatly in 5-HT releaser potency, shown in Table 4. These compounds make ideal tools to explore the interaction of DA and 5-HT release because the DA/5-HT ratios spans >2 orders of magnitude, from 80 for PAL-353 to 1.2 for PAL-313 to 0.27 for PAL-287. Indeed, increasing the potency of 5-HT release had major effects on several behavioral endpoints, including locomotor stimulation in rats (Baumann et al., 2011), reinforcing efficacy in non-human primates (Wee et al., 2005), and food-maintained responding in non-human primates (Negus et al., 2007). The magnitude of behavioral effects correlated with the ratio of DA/5-HT release in all cases, demonstrating that 5-HT release blunts phenotypic DA-mediated behaviors. In vivo microdialysis studies indicate that the levels of extracellular DA decrease as the potency of 5-HT release increases, despite the compounds having similar DA releasing potencies in vitro (Baumann et al., 2011). The two other analogs, PAL-303 and PAL-314, appeared to also correlate with these trends, with locomotor stimulation and extracellular DA levels being increasingly blunted as the level of 5-HT release increases (Baumann et al., 2011). These findings indicate that in vivo the 5-HT system can dampen DA release and DA-mediated behaviors. In the microdialysis studies, extracellular DA concentrations correlate with the extent of locomotor stimulation. PAL-313 with nearly a 1:1 ratio of DA release to 5-HT release in vitro had the highest level of 5-HT release and lowest level of DA release in vivo (Fig. 8). PAL-313 exhibited the lowest levels of locomotor stimulation. The opposite was true for the compound with the highest DA/5-HT release ratio, PAL-353 (Fig. 8). Most importantly, despite the decreases in phenotypic DA-mediated behaviors and decreased levels of extracellular DA, the efficacy of these compounds for decreasing cocaine-maintained responding was not affected. All compounds with equipotent DA and varying 5-HT releasing activity remain efficacious at reducing cocaine self-administration in rhesus monkeys, suggesting that reduced levels of released DA remain therapeutically relevant (Negus et al., 2007). The low levels of DA release found in compounds with high concurrent 5-HT releasing activity suggest that weak non-selective DA releasers may have therapeutic utility with reduced addictive potential.
Table 4. Dopamine (DA) and serotonin (5-HT) releasing effects for compounds that vary in 5-HT releasing potency as determined in rat brain synaptosomes.
| Name | Structure | DA Release EC50 (nM) | 5-HT Release EC50 (nM) | DA/5-HT Ratio |
|---|---|---|---|---|
| PAL-353 |
|
24.2 ± 1.1 | 1937 ± 202 | 80 |
| PAL-303 |
|
51.5 ± 1.7 | 939 ± 76 | 18 |
| PAL-314 |
|
33.3 ± 1.3 | 218 ± 22 | 6.5 |
| PAL-313 |
|
44.1 ± 2.6 | 53.4 ± 4.1 | 1.2 |
| PAL-287 |
|
12.6 ± 0.4 | 3.4 ± 0.2 | 0.27 |
Release EC50 values are reported as means ± SD and are the result of n= 3 performed in triplicate.
Figure 8.
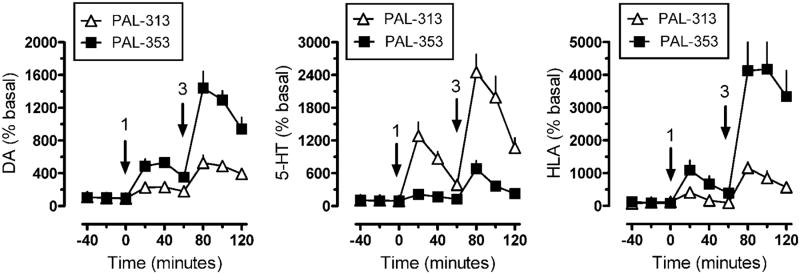
Effects of mixed DAT/SERT releasers on extracellular dopamine (DA), extracellular serotonin (5-HT) and horizontal locomotor activity (HLA) in male rats. Rats undergoing microdialysis in the n. accumbens received sequential intravenous doses of the DAT-selective releaser PAL-353 or the non-selective DAT/SERT releaser PAL-313. Arrows indicate time of injections (1 or 3 mg/kg i.v.). Dialysate DA and 5-HT concentrations were assayed by HPLC-ECD. Data are mean ± SEM for n=6-7 rats/group.
4.5 The Role of VMAT2
One aspect of the release mechanism that remains unclear is how a DA releaser depletes DA from vesicular stores. A DA releaser can induce DA efflux from the cytoplasm via the DAT, but “free” DA molecules must be present in the cytoplasm in order for that process to occur. In vivo microdialysis studies in rats show that amphetamine increases extracellular DA concentrations by acting on non-vesicular and vesicular pools of cytoplasmic transmitter (Butcher et al., 1988; Florin et al., 1995; Watanabe et al., 2005). One hypothesis suggests that releasers deplete DA from vesicles because they are substrates of VMAT2, the primary transporter responsible for uptake of neurotransmitters into vesicles. The action of substrates at VMAT2 appears to be analogous to the effects of substrates on plasma membrane transporters, leading to reversal of normal transporter flux and release of vesicular transmitters into the cytoplasm (Teng et al., 1998; Partilla et al., 2006). The substrate actions at VMAT2 would also inhibit any subsequent DA uptake into vesicles, which serves as an additional mechanism to reduce vesicular transmitter concentrations (see Sulzer, 2011). Indeed, most of the DAT releasers examined to date induce neurotransmitter efflux that involves VMAT2 (Fleckenstein et al., 2007). Another mechanism of vesicular amine depletion is provided by the weak base hypothesis of Sulzer (2011). According to this scenario, substrates such as amphetamine are sufficiently lipophilic to enter vesicles by membrane permeation. Once inside the vesicle interior, the drug molecules increase intracellular pH, which weakens the uptake capability of synaptic vesicles and their ability to retain DA. On the other hand, many observations indicate that dissipation of vesicular pH gradients cannot fully explain the ability of certain compounds to release vesicular DA (Reith and Coffey, 1994; Sulzer, 2011). Most likely, a combination of all the above mechanisms plays a role in vesicular monoamine depletion, with different mechanistic effects for different releaser molecules. It is noteworthy that substrate activity at VMAT2 is not a universal property of DAT releasers. Indeed, several known stimulants have been found to lack VMAT2 activity (Partilla et al., 2006). Phenmetrazine was a DA releasing anorectic therapeutic in the 1950s and early 1960s, sold under the name Predulin® (Rothman et al., 2002) but was removed from the clinic due to its addiction liability. It is a potent DA releaser with an EC50 of 87.4 nM (Table 5). However, phenmetrazine was found to be completely inactive at VMAT2 indicating that a direct interaction of the releaser with VMAT2 is not required for inducing neurotransmitter efflux into the extracellular space (Partilla et al., 2006). Phentermine and benzylpiperazine were also found in the same study to lack VMAT2 activity (Table 5). These compounds thus represent yet another atypical class of releaser. Phenmetrazine has been studied in animal models as a potential therapeutic for stimulant use, both directly and via a clinically approved compound phendimetrazine which acts as a pro-drug for phenmetrazine (Negus et al., 2009; Banks et al., 2013a; 2013b; 2013c). Human laboratory experiments using phendimetrazine are under way.
Table 5. Releasing potency at the dopamine transporter (DAT) and vesicular monoamine transporter 2 (VMAT2) for selected compounds.
| Name | Structure | DAT Release EC50 (nM) | VMAT2 Releasea EC50 (μM) |
|---|---|---|---|
| Amphetamine |
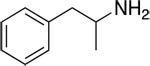
|
8.7 ± 1.2 | 10.5 ± 2.3 |
| PAL-287 |
|
15.7 ± 1 | 1.25 ± 0.23 |
| (+)-Phenmetrazine |
|
87.4 ± 7.8 | >100 |
| Phentermine |
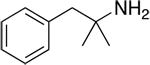
|
262 ± 21 | >100 |
| Benzylpiperazine |
|
307 ± 19 | >100 |
Release EC50 are reported as means ± SD and are the result of n= 3 performed in triplicate.
VMAT2 Release measured with Tyramine.
5. Concluding Remarks
Over the past three decades substantial advances have been made in the understanding of the neurobiological mechanisms of the behavioral and reinforcing effects of cocaine. Though more needs to be accomplished before an avenue to a medical treatment against cocaine abuse becomes clear, many new pharmacological agents targeting the DAT have been discovered, and some of these have effects very different from those producing the abuse liability of cocaine. These atypical compounds have shed some light on mechanisms by which agents may interact with the DAT producing their heterogeneous effects, and have provided new insights into the molecular and behavioral actions of cocaine. Along the way, our understanding of the DAT and transporters in general has been transformed. Initially the DAT was conceptualized not too differently from an energy-driven carrier translocating substrate or a channel through which substrate travelled, for its ultimate intracellular sequestration. The current understanding of the DAT combines the conformational changes of a carrier mechanism with movement of channel-like gates, and such changes in conformation (along with gating) can be altered by ligand which in turn ultimately influences the pharmacology of those ligands.
It is clear from the evidence presented above regarding the interactions of atypical compounds with the DAT, their pharmacokinetic/pharmacodynamic properties, and potential mechanisms involved, that there is no single path to atypical DAT effects. For DAT inhibitors, indicators for likely atypicality are slow onset, combined DAT and σR antagonism, and inward-DAT bias. For partial releasers, a likely indicator for atypicality can be found upon comparing their structure with a closely related substrate (such as amphetamine, dopamine, serotonin, or PAL 1046) and considering a small amount of added molecular size impeding but not fully blocking translocation (the grey area between a substrate and an inhibitor). The current evidence supports multiple types of interaction with the DAT that leads to these atypical pharmacological profiles, and the atypical profiles themselves are not identical. Although modafinil is the only compound that has found its way into clinical testing, it does not mean its particular atypical profile is the only one to focus on for further drug development.
Atypical DAT inhibitors are of interest as potential treatment compounds for stimulant abuse in two areas: compounds can be active as cocaine antagonists or serve as agonist-like substitutes. For monoamine transporter substrates, varying combinations of full and partial releasing ability at different transporters are thought to offer unique therapeutic potentials; and releasing capability at one transporter can be combined with uptake inhibitory activity at another, adding to a panel of potential therapeutic approaches. Moreover, there may as well be additional clinical uses for these compounds in the treatment of ADHD and obesity.
The potential of atypical DAT inhibitors as treatments for ADHD, however, should not restrict drug development solely for treatment of symptoms of impulsivity or self control. Actual drug taking should be kept clearly within the crosshairs of any program with hopes of discovery and development of a useful medication for stimulant abuse. Current institutional emphases on repurposing existing but discarded pharmaceuticals will likely not make significant inroads nor produce a rapid success. Rather, a concerted effort with commensurate funding directed at DAT ligands clearly from the evidence gathered to date (as covered in this Review) shows more promise as an avenue towards candidate treatments for stimulant abuse.
Highlights for review.
Treatment of Stimulant-Use Disorders remains a formidable challenge, and the dopamine transporter (DAT) remains a potential target for antagonist or agonist-like substitution therapies. This review focuses on DAT ligands, such as benztropine, GBR 12909, modafinil, and DAT substrates derived from phenethylamine or cathinone that have atypical DAT-inhibitor effects, either in vitro or in vivo. The compounds are described from a molecular mechanistic, behavioral, and medicinal-chemical perspective.
Mechanisms of atypical DAT inhibitors may serve as targets for the development of treatments for stimulant abuse. These mechanisms are novel and their further exploration may produce compounds with unique therapeutic potential as treatments for stimulant abuse.
Acknowledgments
We thank Dr. Amy Newman for supplies of R- and S-modafinil used to produce results shown in Figure 5.
Role of funding source: The research was supported by National Institutes of Health grants R01 DA019676 (MEAR), MH083840 (MEAR), DA12970 (BEB), as well as travel support from the Behavior, Biology, and Chemistry: Translational Research in Addiction 2014 symposium. Funding for the BBC conference was made possible, in part, by R13DA029347 from the National Institute on Drug Abuse. The research of JLK, WCH, MHB, JSP, and RBH is supported by the NIDA Intramural Research Programs. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Health and Human Services, the National Institute on Drug Abuse, the National Institute of Mental Health, or the University of Texas at San Antonio Health Sciences Center. Mention of trade names, commercial practices, or organization does not imply endorsement by the U.S. Government. The funding sources had no role in writing the review or in the decision to submit the review for publication.
Footnotes
This paper was presented by M.E.A.R., B.E.B., J.L.K. and C.O'B. (see footnote 2) in a symposium at the Behavior, Biology, and Chemistry: Translational Research in Addiction meeting on March 15, 2014 in San Antonio, TX entitled “Non-classical pharmacology of the dopamine transporter and addiction.”
Contributors: MEAR, BEB, and JLK wrote the first draft based on collective conceptual input of all authors at the symposium. All authors edited, substantially revised, are responsible for, and approved the content of the submitted review.
Conflict of interest: The NIH is the owner, and J.L.K. is one of several inventors, on patents covering some of the compounds described in this paper. Temple University and New York University are owners, and M.E.A.R. is one of the two inventors, on patents regarding C-1 cocaine analogs.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adkins EM, Samuvel DJ, Fog JU, Eriksen J, Jayanthi LD, Vaegter CB, Ramamoorthy S, Gether U. Membrane mobility and microdomain association of the dopamine transporter studied with fluorescence correlation spectroscopy and fluorescence recovery after photobleaching. Biochemistry. 2007;46:10484–10497. doi: 10.1021/bi700429z. [DOI] [PubMed] [Google Scholar]
- Anderson AL, Reid MS, Li SH, Holmes T, Shemanski L, Slee A, Smith EV, Kahn R, Chiang N, Vocci F, Ciraulo D, Dackis C, Roache JD, Salloum IM, Somoza E, Urschel HC, III, Elkashef AM. Modafinil for the treatment of cocaine dependence. Drug Alcohol Depend. 2009;104:133–139. doi: 10.1016/j.drugalcdep.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks ML, Blough BE, Fennell TR, Snyder RW, Negus SS. Effects of phendimetrazine treatment on cocaine vs food choice and extended-access cocaine consumption in rhesus monkeys. Neuropsychopharmacol. 2013a;38:2698–2707. doi: 10.1038/npp.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks ML, Blough BE, Fennell TR, Snyder RW, Negus SS. Role of phenmetrazine as an active metabolite of phendimetrazine: evidence from studies of drug discrimination and pharmacokinetics in rhesus monkeys. Drug Alcohol Depend. 2013b;130:158–166. doi: 10.1016/j.drugalcdep.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks ML, Blough BE, Negus SS. Effects of 14-day treatment with the schedule III anorectic phendimetrazine on choice between cocaine and food in rhesus monkeys. Drug Alcohol Depend. 2013c;131:204–213. doi: 10.1016/j.drugalcdep.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AC, Miller JR, Dohrmann JM, Caine SB. Effects of dopamine indirect agonists and selective D1-like and D2-like agonists and antagonists on cocaine self-administration and food maintained responding in rats. Neuropharmacol. 2004;47(Suppl 1):256–273. doi: 10.1016/j.neuropharm.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Baumann MH, Ayestas MA, Dersch CM, Brockington A, Rice KC, Rothman RB. Effects of phentermine and fenfluramine on extracellular dopamine and serotonin in rat nucleus accumbens: therapeutic implications. Synapse. 2000;36:102–113. doi: 10.1002/(SICI)1098-2396(200005)36:2<102::AID-SYN3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Baumann MH, Clark RD, Woolverton WL, Wee S, Blough BE, Rothman RB. In vivo effects of amphetamine analogs reveal evidence for serotonergic inhibition of mesolimbic dopamine transmission in the rat. J Pharmacol Exp Ther. 2011;337:218–225. doi: 10.1124/jpet.110.176271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman J, Madras BK, Johnson SE, Spealman RD. Effects of cocaine and related drugs in nonhuman primates. III Self-administration by squirrel monkeys. J Pharmacol Exp Ther. 1989;251:150–155. [PubMed] [Google Scholar]
- Bergman J, Spealman RD. Some behavioral effects of histamine H1 antagonists in squirrel monkeys. J Pharmacol Exp Ther. 1986;239:104–110. [PubMed] [Google Scholar]
- Blough BE, Landavazo A, Partilla JS, Baumann MH, Decker AM, Page KM, Rothman RB. Hybrid dopamine uptake blocker-serotonin releaser ligands: a new twist on transporter-focused therapeutics. ACS Med Chem Lett. 2014;5:623–627. doi: 10.1021/ml500113s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulling S, Schicker K, Zhang YW, Steinkellner T, Stockner T, Gruber CW, Boehm S, Freissmuth M, Rudnick G, Sitte HH, Sandtner W. The mechanistic basis for noncompetitive ibogaine inhibition of serotonin and dopamine transporters. J Biol Chem. 2012;287:18524–18534. doi: 10.1074/jbc.M112.343681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher SP, Fairbrother IS, Kelly JS, Arbuthnott GW. Amphetamine-induced dopamine release in the rat striatum: an in vivo microdialysis study. J Neurochem. 1988;50:346–355. doi: 10.1111/j.1471-4159.1988.tb02919.x. [DOI] [PubMed] [Google Scholar]
- Campbell VC, Kopajtic TA, Newman AH, Katz JL. Assessment of the influence of histaminergic actions on cocaine-like effects of 3alpha-diphenylmethoxytropane analogs. J Pharmacol Exp Ther. 2005;315:631–640. doi: 10.1124/jpet.105.090829. [DOI] [PubMed] [Google Scholar]
- Cao J, Prisinzano TE, Okunola OM, Kopajtic T, Shook M, Katz JL, Newman AH. Structure-activity relationships at the monoamine transporters for a novel series of modafinil (2-[(diphenylmethyl)sulfinyl]acetamide) analogues. ACS Med Chem Lett. 2010;2:48–52. doi: 10.1021/ml1002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton PL, Didamo P. Augmentation of the behavioral effects of amphetamine by atropine. J Pharmacol Exp Ther. 1961;132:91–96. [PubMed] [Google Scholar]
- Carroll FI, Lewin AH, Mascarella SW. Dopamine transporter uptake blockers. Structure-activity relationships. In: Reith MEA, editor. Neurotransmitter Transporters: Structure, Function, and Regulation. Humana Press; Totowa, NJ: 2002. pp. 381–432. [Google Scholar]
- Carroll FI, Blough BE, Abraham P, Mills AC, Holleman JA, Wolckenhauer SA, Decker AM, Landavazo A, McElroy KT, Navarro HA, Gatch MB, Forster MJ. Synthesis and biological evaluation of bupropion analogues as potential pharmacotherapies for cocaine addiction. J Med Chem. 2009;52:6768–6781. doi: 10.1021/jm901189z. [DOI] [PubMed] [Google Scholar]
- Carroll FI, Pawlush N, Kuhar MJ, Pollard GT, Howard JL. Synthesis, monoamine transporter binding properties, and behavioral pharmacology of a series of 3beta-(substituted phenyl)-2beta-(3′-substituted isoxazol-5-yl)tropanes. J Med Chem. 2004;47:296–302. doi: 10.1021/jm030453p. [DOI] [PubMed] [Google Scholar]
- Cervinski MA, Foster JD, Vaughan RA. Psychoactive substrates stimulate dopamine transporter phosphorylation and down-regulation by cocaine-sensitive and protein kinase C-dependent mechanisms. J Biol Chem. 2005;280:40442–40449. doi: 10.1074/jbc.M501969200. [DOI] [PubMed] [Google Scholar]
- Chen N, Ferrer JV, Javitch JA, Justice JB., Jr Transport-dependent accessibility of a cytoplasmic loop cysteine in the human dopamine transporter. J Biol Chem. 2000;275:1608–1614. doi: 10.1074/jbc.275.3.1608. [DOI] [PubMed] [Google Scholar]
- Chen N, Justice JB., Jr Cocaine acts as an apparent competitive inhibitor at the outward-facing conformation of the human norepinephrine transporter: kinetic analysis of inward and outward transport. J Neurosci. 1998;18:10257–10268. doi: 10.1523/JNEUROSCI.18-24-10257.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Zhen J, Reith MEA. Mutation of Trp84 and Asp313 of the dopamine transporter reveals similar mode of binding interaction for GBR 12909 and benztropine as opposed to cocaine. J Neurochem. 2004;89:853–864. doi: 10.1111/j.1471-4159.2004.02386.x. [DOI] [PubMed] [Google Scholar]
- Cremona ML, Matthies HJ, Pau K, Bowton E, Speed N, Lute BJ, Anderson M, Sen N, Robertson SD, Vaughan RA, Rothman JE, Galli A, Javitch JA, Yamamoto A. Flotillin-1 is essential for PKC-triggered endocytosis and membrane microdomain localization of DAT. Nat Neurosci. 2011;14:469–477. doi: 10.1038/nn.2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dackis CA, Kampman KM, Lynch KG, Pettinati HM, O'Brien CP. A double-blind, placebo-controlled trial of modafinil for cocaine dependence. Neuropsychopharmacol. 2005;30:205–211. doi: 10.1038/sj.npp.1300600. [DOI] [PubMed] [Google Scholar]
- Dackis CA, Kampman KM, Lynch KG, Plebani JG, Pettinati HM, Sparkman T, O'Brien CP. A double-blind, placebo-controlled trial of modafinil for cocaine dependence. J Subst Abuse Treat. 2012;43:303–312. doi: 10.1016/j.jsat.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dackis CA, Lynch KG, Yu E, Samaha FF, Kampman KM, Cornish JW, Rowan A, Poole S, White L, O'Brien CP. Modafinil and cocaine: a double-blind, placebo-controlled drug interaction study. Drug Alcohol Depend. 2003;70:29–37. doi: 10.1016/s0376-8716(02)00335-6. [DOI] [PubMed] [Google Scholar]
- Daws LC, Callaghan PD, Moron JA, Kahlig KM, Shippenberg TS, Javitch JA, Galli A. Cocaine increases dopamine uptake and cell surface expression of dopamine transporters. Biochem Biophys Res Commun. 2002;290:1545–1550. doi: 10.1006/bbrc.2002.6384. [DOI] [PubMed] [Google Scholar]
- Desai RI, Grandy DK, Lupica CR, Katz JL. Pharmacological characterization of a dopamine transporter ligand that functions as a cocaine antagonist. J Pharmacol Exp Ther. 2014;348:106–115. doi: 10.1124/jpet.113.208538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai RI, Kopajtic TA, Koffarnus M, Newman AH, Katz JL. Identification of a dopamine transporter ligand that blocks the stimulant effects of cocaine. J Neurosci. 2005;25:1889–1893. doi: 10.1523/JNEUROSCI.4778-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. New insights into the mechanism of action of amphetamines. Annu Rev Pharmacol Toxicol. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- Florin SM, Kuczenski R, Segal DS. Effects of reserpine on extracellular caudate dopamine and hippocampus norepinephrine responses to amphetamine and cocaine: mechanistic and behavioral considerations. J Pharmacol Exp Ther. 1995;274:231–241. [PubMed] [Google Scholar]
- Foster JD, Adkins SD, Lever JR, Vaughan RA. Phorbol ester induced trafficking-independent regulation and enhanced phosphorylation of the dopamine transporter associated with membrane rafts and cholesterol. J Neurochem. 2008;105:1683–1699. doi: 10.1111/j.1471-4159.2008.05262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach KK, Dasgupta N, Schnoll SH, Henningfield JE. Epidemiology of stimulant misuse and abuse: Implications for future epidemiologic and neuropharmacologic research. Neuropharmacol. 2014;87C:91–96. doi: 10.1016/j.neuropharm.2014.04.020. [DOI] [PubMed] [Google Scholar]
- Gold LH, Balster RL. Evaluation of the cocaine-like discriminative stimulus effects and reinforcing effects of modafinil. Psychopharmacology (Berl) 1996;126:286–292. doi: 10.1007/BF02247379. [DOI] [PubMed] [Google Scholar]
- Gowrishankar R, Hahn MK, Blakely RD. Good riddance to dopamine: roles for the dopamine transporter in synaptic function and dopamine-associated brain disorders. Neurochem Int. 2014;73:42–48. doi: 10.1016/j.neuint.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Grabowski J, Rhoades H, Schmitz J, Stotts A, Daruzska LA, Creson D, Moeller FG. Dextroamphetamine for cocaine-dependence treatment: a double-blind randomized clinical trial. J Clin Psychopharmacol. 2001;21:522–526. doi: 10.1097/00004714-200110000-00010. [DOI] [PubMed] [Google Scholar]
- Grabowski J, Shearer J, Merrill J, Negus SS. Agonist-like, replacement pharmacotherapy for stimulant abuse and dependence. Addict Behav. 2004;29:1439–1464. doi: 10.1016/j.addbeh.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gundlah C, Hjorth S, Auerbach SB. Autoreceptor antagonists enhance the effect of the reuptake inhibitor citalopram on extracellular 5-HT: this effect persists after repeated citalopram treatment. Neuropharmacol. 1997;36:475–482. doi: 10.1016/s0028-3908(97)00052-x. [DOI] [PubMed] [Google Scholar]
- Haile CN, Zhang XY, Carroll FI, Kosten TA. Cocaine self-administration and locomotor activity are altered in Lewis and F344 inbred rats by RTI 336, a 3-phenyltropane analog that binds to the dopamine transporter. Brain Res. 2005;1055:186–195. doi: 10.1016/j.brainres.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Hart CL, Haney M, Vosburg SK, Rubin E, Foltin RW. Smoked cocaine self-administration is decreased by modafinil. Neuropsychopharmacol. 2008;33:761–768. doi: 10.1038/sj.npp.1301472. [DOI] [PubMed] [Google Scholar]
- Hiranita T, Kohut SJ, Soto PL, Tanda G, Kopajtic TA, Katz JL. Preclinical efficacy of N-substituted benztropine analogs as antagonists of methamphetamine self-administration in rats. J Pharmacol Exp Ther. 2014;348:174–191. doi: 10.1124/jpet.113.208264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiranita T, Soto PL, Kohut SJ, Kopajtic T, Cao J, Newman AH, Tanda G, Katz JL. Decreases in cocaine self-administration with dual inhibition of the dopamine transporter and sigma receptors. J Pharmacol Exp Ther. 2011;339:662–677. doi: 10.1124/jpet.111.185025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiranita T, Soto PL, Newman AH, Katz JL. Assessment of reinforcing effects of benztropine analogs and their effects on cocaine self-administration in rats: comparisons with monoamine uptake inhibitors. J Pharmacol Exp Ther. 2009;329:677–686. doi: 10.1124/jpet.108.145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiranita T, Wilkinson DS, Hong WC, Zou MF, Kopajtic TA, Soto PL, Lupica CR, Newman AH, Katz JL. 2-isoxazol-3-phenyltropane derivatives of cocaine: molecular and atypical system effects at the dopamine transporter. J Pharmacol Exp Ther. 2014;349:297–309. doi: 10.1124/jpet.113.212738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong WC, Amara SG. Membrane cholesterol modulates the outward-facing conformation of the dopamine transporter and alters cocaine binding. J Biol Chem. 2010;285:32616–32626. doi: 10.1074/jbc.M110.150565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell LL, Carroll FI, Votaw JR, Goodman MM, Kimmel HL. Effects of combined dopamine and serotonin transporter inhibitors on cocaine self-administration in rhesus monkeys. J Pharmacol Exp Ther. 2007;320:757–765. doi: 10.1124/jpet.106.108324. [DOI] [PubMed] [Google Scholar]
- Howell LL, Negus SS. Monoamine transporter inhibitors and substrates as treatments for stimulant abuse. Adv Pharmacol. 2014;69:129–176. doi: 10.1016/B978-0-12-420118-7.00004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izenwasser S, Newman AH, Katz JL. Cocaine and several sigma receptor ligands inhibit dopamine uptake in rat caudate-putamen. Eur J Pharmacol. 1993;243:201–205. doi: 10.1016/0014-2999(93)90381-q. [DOI] [PubMed] [Google Scholar]
- Jacobs MT, Zhang YW, Campbell SD, Rudnick G. Ibogaine, a noncompetitive inhibitor of serotonin transport, acts by stabilizing the cytoplasm-facing state of the transporter. J Biol Chem. 2007;282:29441–29447. doi: 10.1074/jbc.M704456200. [DOI] [PubMed] [Google Scholar]
- Jones KT, Zhen J, Reith MEA. Importance of cholesterol in dopamine transporter function. J Neurochem. 2012;123:700–715. doi: 10.1111/jnc.12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karila L, Reynaud M. Therapeutic approaches to cocaine addiction. Rev Prat. 2009;59:830–834. [PubMed] [Google Scholar]
- Katz JL, Agoston GE, Alling KL, Kline RH, Forster MJ, Woolverton WL, Kopajtic TA, Newman AH. Dopamine transporter binding without cocaine-like behavioral effects: synthesis and evaluation of benztropine analogs alone and in combination with cocaine in rodents. Psychopharmacology (Berl) 2001;154:362–374. doi: 10.1007/s002130000667. [DOI] [PubMed] [Google Scholar]
- Katz JL, Izenwasser S, Kline RH, Allen AC, Newman AH. Novel 3alpha-diphenylmethoxytropane analogs: selective dopamine uptake inhibitors with behavioral effects distinct from those of cocaine. J Pharmacol Exp Ther. 1999a;288:302–315. [PubMed] [Google Scholar]
- Katz JL, Izenwasser S, Kline RH, Allen AC, Newman AH. Novel 3alpha-diphenylmethoxytropane analogs: selective dopamine uptake inhibitors with behavioral effects distinct from those of cocaine. J Pharmacol Exp Ther. 1999b;288:302–315. [PubMed] [Google Scholar]
- Katz JL, Izenwasser S, Terry P. Relationships among dopamine transporter affinities and cocaine-like discriminative-stimulus effects. Psychopharmacology (Berl) 2000;148:90–98. doi: 10.1007/s002130050029. [DOI] [PubMed] [Google Scholar]
- Katz JL, Kopajtic TA, Agoston GE, Newman AH. Effects of N-substituted analogs of benztropine: diminished cocaine-like effects in dopamine transporter ligands. J Pharmacol Exp Ther. 2004;309:650–660. doi: 10.1124/jpet.103.060525. [DOI] [PubMed] [Google Scholar]
- Katz JL, Su TP, Hiranita T, Hayashi T, Tanda G, Kopajtic T, Tsai SY. A Role for sigma receptors in stimulant self administration and addiction. Pharmaceuticals (Basel) 2011;4:880–914. doi: 10.3390/ph4060880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004;2:E78. doi: 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A. Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem. 2003;278:12070–12077. doi: 10.1074/jbc.M212815200. [DOI] [PubMed] [Google Scholar]
- Kitayama S, Shimada S, Xu H, Markham L, Donovan DM, Uhl GR. Dopamine transporter site-directed mutations differentially alter substrate transport and cocaine binding. Proc Natl Acad Sci U S A. 1992;89:7782–7785. doi: 10.1073/pnas.89.16.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohut SJ, Hiranita T, Hong SK, Ebbs AL, Tronci V, Green J, Garces-Ramirez L, Chun LE, Mereu M, Newman AH, Katz JL, Tanda G. Preference for Distinct functional conformations of the dopamine transporter alters the relationship between subjective effects of cocaine and stimulation of mesolimbic dopamine. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.03.031. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopajtic TA, Liu Y, Surratt CK, Donovan DM, Newman AH, Katz JL. Dopamine transporter-dependent and -independent striatal binding of the benztropine analog JHW 007, a cocaine antagonist with low abuse liability. J Pharmacol Exp Ther. 2010;335:703–714. doi: 10.1124/jpet.110.171629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481:469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leviel V. Dopamine release mediated by the dopamine transporter, facts and consequences. J Neurochem. 2011;118:475–489. doi: 10.1111/j.1471-4159.2011.07335.x. [DOI] [PubMed] [Google Scholar]
- Li SM, Kopajtic TA, O'Callaghan MJ, Agoston GE, Cao J, Newman AH, Katz JL. N-substituted benztropine analogs: selective dopamine transporter ligands with a fast onset of action and minimal cocaine-like behavioral effects. J Pharmacol Exp Ther. 2011;336:575–585. doi: 10.1124/jpet.110.173260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llosa T, Llosa L. Handbook on oral cocaine as an agonist therapy in cocaine dependence. [last accessed 10-15-2014];Public Domain. 2005 http://www.bvcedro.org.pe/bitstream/123456789/415/1/833-BRCM-S-COCA.pdf.
- Loland CJ, Desai RI, Zou MF, Cao J, Grundt P, Gerstbrein K, Sitte HH, Newman AH, Katz JL, Gether U. Relationship between conformational changes in the dopamine transporter and cocaine-like subjective effects of uptake inhibitors. Mol Pharmacol. 2008;73:813–823. doi: 10.1124/mol.107.039800. [DOI] [PubMed] [Google Scholar]
- Loland CJ, Mereu M, Okunola OM, Cao J, Prisinzano TE, Mazier S, Kopajtic T, Shi L, Katz JL, Tanda G, Newman AH. R-modafinil (armodafinil): a unique dopamine uptake inhibitor and potential medication for psychostimulant abuse. Biol Psychiatry. 2012;72:405–413. doi: 10.1016/j.biopsych.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolm R, Swayngim K, Donovan JL, DeVane CL, Elkashef A, Chiang N, Khan R, Mojsiak J, Myrick DL, Hedden S, Cochran K, Woolson RF. Modafinil and cocaine interactions. Am J Drug Alcohol Abuse. 2006;32:577–587. doi: 10.1080/00952990600920425. [DOI] [PubMed] [Google Scholar]
- Martin-Fardon R, Maurice T, Aujla H, Bowen WD, Weiss F. Differential effects of sigma1 receptor blockade on self-administration and conditioned reinstatement motivated by cocaine vs natural reward. Neuropsychopharmacol. 2007;32:1967–1973. doi: 10.1038/sj.npp.1301323. [DOI] [PubMed] [Google Scholar]
- Matsumoto RR. Targeting sigma receptors: novel medication development for drug abuse and addiction. Expert Rev Clin Pharmacol. 2009;2:351–358. doi: 10.1586/ecp.09.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello NK, Negus SS. Effects of d-amphetamine and buprenorphine combinations on speedball (cocaine+heroin) self-administration by rhesus monkeys. Neuropsychopharmacol. 2007;32:1985–1994. doi: 10.1038/sj.npp.1301319. [DOI] [PubMed] [Google Scholar]
- Mereu M, Bonci A, Newman AH, Tanda G. The neurobiology of modafinil as an enhancer of cognitive performance and a potential treatment for substance use disorders. Psychopharmacology (Berl) 2013;229:415–434. doi: 10.1007/s00213-013-3232-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minzenberg MJ, Carter CS. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacol. 2008;33:1477–1502. doi: 10.1038/sj.npp.1301534. [DOI] [PubMed] [Google Scholar]
- Mooney ME, Herin DV, Schmitz JM, Moukaddam N, Green CE, Grabowski J. Effects of oral methamphetamine on cocaine use: a randomized, double-blind, placebo-controlled trial. Drug Alcohol Depend. 2009;101:34–41. doi: 10.1016/j.drugalcdep.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro HA, Howard JL, Pollard GT, Carroll FI. Positive allosteric modulation of the human cannabinoid (CB) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br J Pharmacol. 2009;156:1178–1184. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Baumann MH, Rothman RB, Mello NK, Blough BE. Selective suppression of cocaine- versus food-maintained responding by monoamine releasers in rhesus monkeys: benzylpiperazine, (+)phenmetrazine, and 4-benzylpiperidine. J Pharmacol Exp Ther. 2009;329:272–281. doi: 10.1124/jpet.108.143701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Mello NK. Effects of chronic d-amphetamine treatment on cocaine- and food-maintained responding under a second-order schedule in rhesus monkeys. Drug Alcohol Depend. 2003;70:39–52. doi: 10.1016/s0376-8716(02)00339-3. [DOI] [PubMed] [Google Scholar]
- Negus SS, Mello NK, Blough BE, Baumann MH, Rothman RB. Monoamine releasers with varying selectivity for dopamine/norepinephrine versus serotonin release as candidate “agonist” medications for cocaine dependence: studies in assays of cocaine discrimination and cocaine self-administration in rhesus monkeys. J Pharmacol Exp Ther. 2007;320:627–636. doi: 10.1124/jpet.106.107383. [DOI] [PubMed] [Google Scholar]
- Newman JL, Negus SS, Lozama A, Prisinzano TE, Mello NK. Behavioral evaluation of modafinil and the abuse-related effects of cocaine in rhesus monkeys. Exp Clin Psychopharmacol. 2010;18:395–408. doi: 10.1037/a0021042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien CP. Modafinil-cocaine interactions: clinical implications. Biol Psychiatry. 2012;72:346. doi: 10.1016/j.biopsych.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Partilla JS, Dempsey AG, Nagpal AS, Blough BE, Baumann MH, Rothman RB. Interaction of amphetamines and related compounds at the vesicular monoamine transporter. J Pharmacol Exp Ther. 2006;319:237–246. doi: 10.1124/jpet.106.103622. [DOI] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KA, Epstein DH, Preston KL. Psychostimulant addiction treatment. Neuropharmacol. 2014 Apr 12; doi: 10.1016/j.neuropharm.2014.04.002. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, Di Chiara G. Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the shell as compared with the core of the rat nucleus accumbens. Proc Natl Acad Sci U S A. 1995;92:12304–12308. doi: 10.1073/pnas.92.26.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Shi L, Zehnpfennig B, Weinstein H, Javitch JA. Experimental conditions can obscure the second high-affinity site in LeuT. Nat Struct Mol Biol. 2012;19:207–211. doi: 10.1038/nsmb.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raje S, Cao J, Newman AH, Gao H, Eddington ND. Evaluation of the blood-brain barrier transport, population pharmacokinetics, and brain distribution of benztropine analogs and cocaine using in vitro and in vivo techniques. J Pharmacol Exp Ther. 2003;307:801–808. doi: 10.1124/jpet.103.053504. [DOI] [PubMed] [Google Scholar]
- Rea WP, Rothman RB, Shippenberg TS. Evaluation of the conditioned reinforcing effects of phentermine and fenfluramine in the rat: concordance with clinical studies. Synapse. 1998;30:107–111. doi: 10.1002/(SICI)1098-2396(199809)30:1<107::AID-SYN13>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Reith MEA, Ali S, Hashim A, Sheikh IS, Theddu N, Gaddiraju NV, Mehrotra S, Schmitt KC, Murray TF, Sershen H, Unterwald EM, Davis FS. Novel C-1 substituted cocaine analogues unlike cocaine or benztropine. J Pharmacol Exp Ther. 2012;343:413–425. doi: 10.1124/jpet.112.193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith MEA, Berfield JL, Wang LC, Ferrer JV, Javitch JA. The uptake inhibitors cocaine and benztropine differentially alter the conformation of the human dopamine transporter. J Biol Chem. 2001;276:29012–29018. doi: 10.1074/jbc.M011785200. [DOI] [PubMed] [Google Scholar]
- Reith MEA, Coffey LL. Structure-activity relationships for cocaine congeners in inhibiting dopamine uptake into rat brain synaptic vesicles and bovine chromaffin granule ghosts. J Pharmacol Exp Ther. 1994;271:1444–1452. [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH. Therapeutic and adverse actions of serotonin transporter substrates. Pharmacol Ther. 2002;95:73–88. doi: 10.1016/s0163-7258(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Blough BE, Jacobson AE, Rice KC, Partilla JS. Evidence for noncompetitive modulation of substrate-induced serotonin release. Synapse. 2010;64:862–869. doi: 10.1002/syn.20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Becketts KM, Radesca LR, De Costa BR, Rice KC, Carroll FI, Dersch CM. Studies of the biogenic amine transporters. II. A brief study on the use of [3H]DA-uptake-inhibition to transporter-binding-inhibition ratios for the in vitro evaluation of putative cocaine antagonists. Life Sci. 1993;53:L267–L272. doi: 10.1016/0024-3205(93)90602-y. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Blough BE, Woolverton WL, Anderson KG, Negus SS, Mello NK, Roth BL, Baumann MH. Development of a rationally designed, low abuse potential, biogenic amine releaser that suppresses cocaine self-administration. J Pharmacol Exp Ther. 2005;313:1361–1369. doi: 10.1124/jpet.104.082503. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Elmer GI, Shippenberg TS, Rea W, Baumann MH. Phentermine and fenfluramine. Preclinical studies in animal models of cocaine addiction. Ann N Y Acad Sci. 1998;844:59–74. [PubMed] [Google Scholar]
- Rothman RB, Katsnelson M, Vu N, Partilla JS, Dersch CM, Blough BE, Baumann MH. Interaction of the anorectic medication, phendimetrazine, and its metabolites with monoamine transporters in rat brain. Eur J Pharmacol. 2002;447:51–57. doi: 10.1016/s0014-2999(02)01830-7. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Partilla JS, Baumann MH, Lightfoot-Siordia C, Blough BE. Studies of the biogenic amine transporters. 14. Identification of low-efficacy “partial” substrates for the biogenic amine transporters. J Pharmacol Exp Ther. 2012;341:251–262. doi: 10.1124/jpet.111.188946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckel CL, Boff E. Behavioral effects of interacting imipramine and other drugs with d-amphetamine, cocaine, and tetrabenazine. Psychopharmacologia. 1964;5:198–208. doi: 10.1007/BF00413242. [DOI] [PubMed] [Google Scholar]
- Schenk S. Effects of GBR 12909, WIN 35,428 and indatraline on cocaine self-administration and cocaine seeking in rats. Psychopharmacology (Berl) 2002;160:263–270. doi: 10.1007/s00213-001-0972-3. [DOI] [PubMed] [Google Scholar]
- Schmitt KC, Reith MEA. The atypical stimulant and nootropic modafinil interacts with the dopamine transporter in a different manner than classical cocaine-like inhibitors. PLoS One. 2011;6:e25790. doi: 10.1371/journal.pone.0025790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt KC, Rothman RB, Reith MEA. Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators, and partial substrates. J Pharmacol Exp Ther. 2013;346:2–10. doi: 10.1124/jpet.111.191056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddik A, Holy M, Weissensteiner R, Zdrazil B, Sitte HH, Ecker GF. Probing the selectivity of monoamine transporter substrates by means of molecular modeling. Mol Inform. 2013;32:409–413. doi: 10.1002/minf.201300013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinsen K, Kraft JF, Koldso H, Vinberg KA, Rothman RB, Partilla JS, Wiborg O, Blough B, Schiott B, Sinning S. Binding of the amphetamine-like 1-phenyl-piperazine to monoamine transporters. ACS Chem Neurosci. 2012;3:693–705. doi: 10.1021/cn300040f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- Sitte HH, Freissmuth M. The reverse operation of Na(+)/Cl(-)-coupled neurotransmitter transporters--why amphetamines take two to tango. J Neurochem. 2010;112:340–355. doi: 10.1111/j.1471-4159.2009.06474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitte HH, Huck S, Reither H, Boehm S, Singer EA, Pifl C. Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter. J Neurochem. 1998;71:1289–1297. doi: 10.1046/j.1471-4159.1998.71031289.x. [DOI] [PubMed] [Google Scholar]
- Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J Neurosci. 1997;17:960–974. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Tanda G. Modulation of the endocannabinoid system: therapeutic potential against cocaine dependence. Pharmacol Res. 2007;56:406–417. doi: 10.1016/j.phrs.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanda G, Ebbs A, Newman AH, Katz JL. Effects of 4′-chloro-3 alpha-(diphenylmethoxy)-tropane on mesostriatal, mesocortical, and mesolimbic dopamine transmission: comparison with effects of cocaine. J Pharmacol Exp Ther. 2005;313:613–620. doi: 10.1124/jpet.104.080465. [DOI] [PubMed] [Google Scholar]
- Tanda G, Ebbs AL, Kopajtic TA, Elias LM, Campbell BL, Newman AH, Katz JL. Effects of muscarinic M1 receptor blockade on cocaine-induced elevations of brain dopamine levels and locomotor behavior in rats. J Pharmacol Exp Ther. 2007;321:334–344. doi: 10.1124/jpet.106.118067. [DOI] [PubMed] [Google Scholar]
- Tanda G, Kopajtic TA, Katz JL. Cocaine-like neurochemical effects of antihistaminic medications. J Neurochem. 2008;106:147–157. doi: 10.1111/j.1471-4159.2008.05361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanda G, Li SM, Mereu M, Thomas AM, Ebbs AL, Chun LE, Tronci V, Green JL, Zou MF, Kopajtic TA, Newman AH, Katz JL. Relations between stimulation of mesolimbic dopamine and place conditioning in rats produced by cocaine or drugs that are tolerant to dopamine transporter conformational change. Psychopharmacology (Berl) 2013;229:307–321. doi: 10.1007/s00213-013-3109-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanda G, Newman AH, Katz JL. Discovery of drugs to treat cocaine dependence: behavioral and neurochemical effects of atypical dopamine transport inhibitors. Adv Pharmacol. 2009;57:253–289. doi: 10.1016/S1054-3589(08)57007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanda G, Pontieri FE, Di CG. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science. 1997;276:2048–2050. doi: 10.1126/science.276.5321.2048. [DOI] [PubMed] [Google Scholar]
- Teng L, Crooks PA, Dwoskin LP. Lobeline displaces [3H]dihydrotetrabenazine binding and releases [3H]dopamine from rat striatal synaptic vesicles: comparison with d-amphetamine. J Neurochem. 1998;71:258–265. doi: 10.1046/j.1471-4159.1998.71010258.x. [DOI] [PubMed] [Google Scholar]
- Tuomisto J, Tuomisto L. Effects of histamine and histamine antagonists on the uptake and release of catecholamines and 5-HT in brain synaptosomes. Med Biol. 1980;58:33–37. [PubMed] [Google Scholar]
- Ukairo OT, Bondi CD, Newman AH, Kulkarni SS, Kozikowski AP, Pan S, Surratt CK. Recognition of benztropine by the dopamine transporter (DAT) differs from that of the classical dopamine uptake inhibitors cocaine, methylphenidate, and mazindol as a function of a DAT transmembrane 1 aspartic acid residue. J Pharmacol Exp Ther. 2005;314:575–583. doi: 10.1124/jpet.105.085829. [DOI] [PubMed] [Google Scholar]
- Valchar M, Hanbauer I. Comparison of [3H]WIN 35,428 binding, a marker for dopamine transporter, in embryonic mesencephalic neuronal cultures with striatal membranes of adult rats. J Neurochem. 1993;60:469–476. doi: 10.1111/j.1471-4159.1993.tb03174.x. [DOI] [PubMed] [Google Scholar]
- Vosburg SK, Hart CL, Haney M, Rubin E, Foltin RW. Modafinil does not serve as a reinforcer in cocaine abusers. Drug Alcohol Depend. 2010;106:233–236. doi: 10.1016/j.drugalcdep.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Goehring A, Wang KH, Penmatsa A, Ressler R, Gouaux E. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature. 2013;503:141–145. doi: 10.1038/nature12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Aono Y, Fusa K, Takada K, Saigusa T, Koshikawa N, Cools AR. Contribution of vesicular and cytosolic dopamine to the increased striatal dopamine efflux elicited by intrastriatal injection of dexamphetamine. Neuroscience. 2005;136:251–257. doi: 10.1016/j.neuroscience.2005.07.041. [DOI] [PubMed] [Google Scholar]
- Wee S, Anderson KG, Baumann MH, Rothman RB, Blough BE, Woolverton WL. Relationship between the serotonergic activity and reinforcing effects of a series of amphetamine analogs. J Pharmacol Exp Ther. 2005;313:848–854. doi: 10.1124/jpet.104.080101. [DOI] [PubMed] [Google Scholar]
- Wojnicki FH, Rothman RB, Rice KC, Glowa JR. Effects of phentermine on responding maintained under multiple fixed-ratio schedules of food and cocaine presentation in the rhesus monkey. J Pharmacol Exp Ther. 1999;288:550–560. [PubMed] [Google Scholar]
- Wong EH, Sonders MS, Amara SG, Tinholt PM, Piercey MF, Hoffmann WP, Hyslop DK, Franklin S, Porsolt RD, Bonsignori A, Carfagna N, McArthur RA. Reboxetine: a pharmacologically potent, selective, and specific norepinephrine reuptake inhibitor. Biol Psychiatry. 2000;47:818–829. doi: 10.1016/s0006-3223(99)00291-7. [DOI] [PubMed] [Google Scholar]
- Woolverton WL, Hecht GS, Agoston GE, Katz JL, Newman AH. Further studies of the reinforcing effects of benztropine analogs in rhesus monkeys. Psychopharmacology (Berl) 2001;154:375–382. doi: 10.1007/s002130000616. [DOI] [PubMed] [Google Scholar]
- Woolverton WL, Rowlett JK, Wilcox KM, Paul IA, Kline RH, Newman AH, Katz JL. 3′- and 4′-chloro-substituted analogs of benztropine: intravenous self-administration and in vitro radioligand binding studies in rhesus monkeys. Psychopharmacology (Berl) 2000 Jan;147:426–435. doi: 10.1007/s002130050012. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Yu H, Rothman RB, Dersch CM, Partilla JS, Rice KC. Uptake and release effects of diethylpropion and its metabolites with biogenic amine transporters. Bioorg Med Chem. 2000;8:2689–2692. doi: 10.1016/s0968-0896(00)00210-8. [DOI] [PubMed] [Google Scholar]
- Zahniser NR, Sorkin A. Rapid regulation of the dopamine transporter: role in stimulant addiction? Neuropharmacol. 2004;47(Suppl 1):80–91. doi: 10.1016/j.neuropharm.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith MEA, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith MEA, Wang DN. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat Struct Mol Biol. 2009;16:652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou MF, Kopajtic T, Katz JL, Newman AH. Structure-activity relationship comparison of (S)-2beta-substituted 3alpha-(bis[4-fluorophenyl]methoxy)tropanes and (R)-2beta-substituted 3beta-(3,4-dichlorophenyl)tropanes at the dopamine transporter. J Med Chem. 2003;46:2908–2916. doi: 10.1021/jm0300375. [DOI] [PubMed] [Google Scholar]